The decline in mitochondrial function resulting from aging is known to cause a range of diseases, including stroke, neurodegenerative disorders, cardiomyopathy, and cancer. This relationship between mitochondria and aging is a key topic in research focused on healthy life expectancy. One of the many and varied impacts of aging is its connection to autophagy, the mechanism whereby old organelles and proteins are broken down. A decline in autophagy leads to the accumulation of aged mitochondria, resulting in cellular damage. Attention is focusing on the existence of substances that delay this functional decline.
Special Feature 1 – The Impact of Mitochondria Aiming to extend healthy life expectancy by delaying age-related functional decline
composition by Takeaki Kikuchi

Mikako Yagi
Assistant Professor, Division of Biological Science and Technology, Department of Health Sciences, Graduate School of Medical Sciences, Kyushu University
Graduated from the Department of Biotechnology at Ritsumeikan University’s Faculty of Science and Engineering in 2000. In 2002, she completed a master’s program in the Department of Genetic Resources Technology at Kyushu University’s Graduate School of Bioresource and Bioenvironmental Sciences. In 2003, she became a research fellow in the Department of Clinical Chemistry and Laboratory Medicine at Kyushu University’s Faculty of Medical Sciences, from which she obtained her Ph.D. in 2013. She has held her current position since 2019.
The eukaryotic cells in mammalian bodies contain not only a nucleus, which encloses genes within the nuclear membrane and manages genetic information, but also a number of structures called organelles, which include mitochondria, the Golgi apparatus (organelles that transport proteins by binding them to sugars), and lysosomes (organelles that break down unnecessary substances and waste products in the cell). Of these, mitochondria specialize in producing energy for biological activity.
Mitochondrial function declines due to aging
The genetic information in the mitochondrial DNA (mtDNA) present in mitochondria is transcribed into messenger RNA (mRNA) and translated within the mitochondria. Failure of this translation mechanism has adverse impacts on organs with a large number of mitochondria, such as the brain, heart, and muscles, which can result in a variety of conditions referred to as mitochondrial diseases, including stroke, cerebral ataxia, cardiomyopathy, external ophthalmoplegia, and weakness of the muscles in the limbs.
If mitochondria become dysfunctional, nuclear DNA is also affected, impairing the mechanism whereby lysosomes—which could be described as the recycling centers of cells—break down unnecessary substances, damaged organelles, and waste products.
By engaging in crosstalk and rigorously controlling each other, the mitochondria, cell nucleus, and lysosomes maintain homeostasis within the cell. The relationship between these three elements is also connected to the link between mitochondria and aging.
We know that, generally speaking, mitochondrial function declines with age. As this decline progresses, what was formerly only a very low level of mitochondrial dysfunction gradually increases. And we already know that this growing dysfunction is accompanied by the onset of metabolic syndrome, neurodegenerative disorders, cardiomyopathy, and cancer. More recently, it has been scientifically proven that declines in autophagy (the process in which cells break down and recycle substances such as their old proteins and organelles using lysosomes) and decline in the level of nicotinamide adenine dinucleotide (NAD+) in the blood are also related to aging.
Allow me to provide a simple explanation of autophagy. When a cell is in a state of starvation, a vesicle called a phagophore, or isolation membrane, appears in the cytoplasm. This membrane envelops unnecessary substances in the cell. Once sealed, it is called an autophagosome. Autophagosomes contain mitochondria and other organelles. The autophagosomes then fuse with lysosomes, which break down their contents. We are conducting research into the degradation mechanism involved in autophagy, with a particular focus on the lysosomes with which autophagosomes fuse.
As NAD+ activates energy production within the body, it is believed that it has a diverse array of effects, such as improving cellular function, delaying aging, enhancing cognitive function and exercise capacity, maintaining skin elasticity and luster, and boosting immune function. Reduced autophagy function and NAD+ levels are known to be causes of age-related diseases. This coenzyme has been attracting increasing attention of late; in particular, research into the involvement of NAD+ and sirtuin genes—a gene cluster that could potentially extend lifespan by curbing cellular aging—has been progressing, with research indicating that the view that NAD+ supplementation is required to activate the sirtuin genes.
Which organs are affected by age-related mitochondrial dysfunction and in what way varies from one person to another. Accordingly, the organs subject to associated functional decline in the form of age-related diseases also differ according to the individual. We focused on the heart as an organ affected by aging. In order to investigate the relationship between reduced mitochondrial function in heart tissue and autophagy, particularly lysosomal function, we conducted an exhaustive analysis of cardiac tissue using a method called metabolomic analysis. As a result, we discovered that NAD+ levels decrease. We are now moving forward with a study focused on why NAD+ levels decrease and how we can prevent this decline.
Autophagic degradation activity also decreases
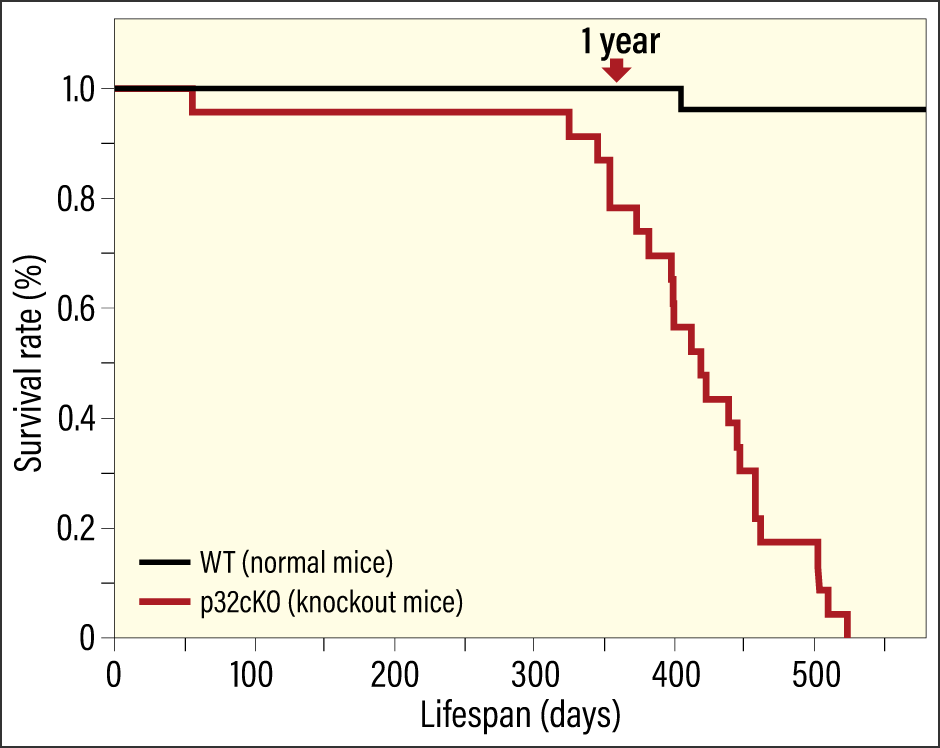
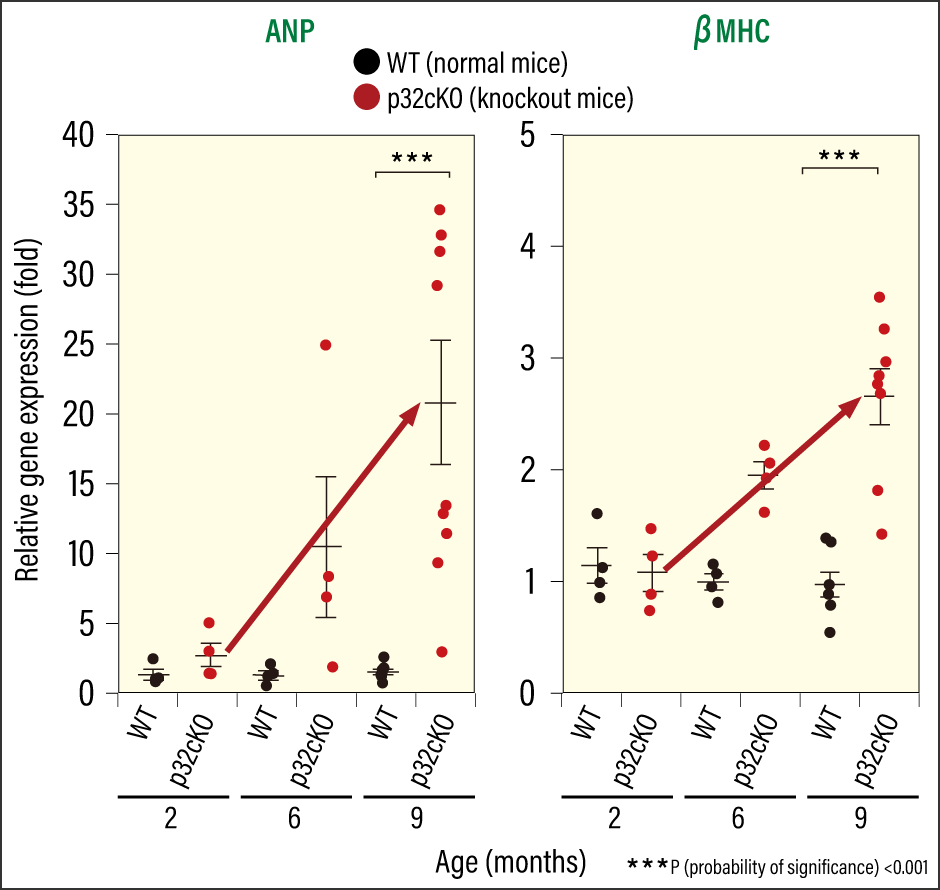
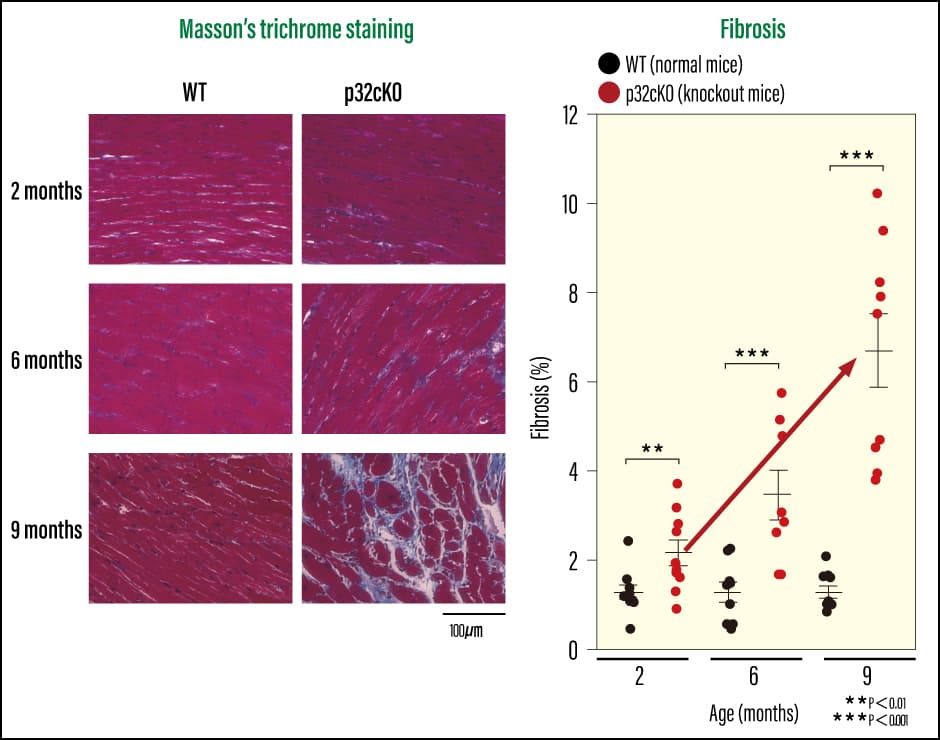
Cardiomyocyte-specific p32 conditional knockout mice (p32cKO)—mice in which the molecule responsible for protein translation in mitochondria, the protein p32, had been removed—had a shorter lifespan than normal mice (which have a lifespan of around two years), at just one year when plotted on a survival curve (Figure 1). When we used ultrasound to check the cardiac function of these mice, we found that they had poor left ventricular ejection fraction and left ventricular fractional shortening. This means that cardiac systolic function was reduced, indicating a strong possibility of early-onset dilated cardiomyopathy. When we examined gene expression in heart failure markers (ANP and βMHC), and investigated cardiac fibrosis using Masson’s trichrome staining, we confirmed that the knockout mice did indeed have reduced cardiac function (Figures 2 and 3). We also found that mitochondrial translation was impaired in the cardiac tissue, leading to reduced protein expression. In other words, the loss of a single molecule that plays an important role in mitochondrial translation reduced cardiac function and shortened lifespan.
 Yagi M. et al: Cardiovascular Research 2017:113 1173–85
Yagi M. et al: Cardiovascular Research 2017:113 1173–85
Figure 1. Survival curve for cardiomyocyte-specific p32cKOThe survival rate of cardiomyocyte-specific p32cKO declines sharply after approximately 300 days. Unlike normal mice, their lifespan is only about a year.
 Yagi M. et al :Cardiovascular Research 2017:113 1173-85
Yagi M. et al :Cardiovascular Research 2017:113 1173-85
Figure 2. Changes in gene expression of heart failure markersThe research team investigated gene expression in the heart failure markers ANP and βMHC. They discovered that gene expression in knockout mice increased over time, indicating progressive deterioration of cardiac function.
 Yagi M. et al: Cardiovascular Research 2017:113 1173–85
Yagi M. et al: Cardiovascular Research 2017:113 1173–85
Figure 3. Assessment of cardiac fibrosisWhen the research team assessed cardiac fibrosis using Masson’s trichrome staining (MT), they found that fibrosis began to occur in the knockout mice from the age of two months, and that the fibrotic area progressively increased thereafter.
First of all, mitochondrial translation was impaired in the cardiac tissue of these mice. This resulted in a rise in expression of the HIF-1α transcription factor (a protein that regulates gene expression) in the nucleus. We know that HIF-1α is one of the first transcription factors to be expressed when an abnormality occurs in a cell, and that it is known to be involved in a variety of diseases, including cancer and ischemic heart disease. We believe that the rise in HIF-1α suppressed the expression of Nmnat3, the enzyme that synthesizes NAD+, causing NAD+ levels to decline. Because of this, the lysosomal degradative activity mediated by autophagy also fell. Cardiac function thus really did decline as a result of crosstalk between mitochondria, the cell nucleus, and lysosomes.
The lysosomes of the knockout mice were swollen compared with those of ordinary mice. This suggests that the lysosomes that ought to break down unnecessary substances have a buildup of substances that cannot be degraded.
Accordingly, we investigated what these substances might be. As it is difficult to conduct experiments in living mice, we carried out repeated experiments in cultured cells. From the results, we discovered that iron in particular had accumulated within lysosomes due to mitochondrial dysfunction. We also confirmed a buildup of lipid peroxides. This damages cellular and organelle membranes, causing damage to cells. This phenomenon is known to induce ferroptosis, an iron-dependent form of cell death. In other words, it is likely that ferroptosis was induced, causing cardiac function to decline and shortening the lifespan of the mice.
Substances involved in the production of energy
Normally, we have to be careful to ensure we are not iron-deficient, as this can cause symptoms of anemia, including fatigue, shortness of breath, and dizziness. Women, especially, are told to ensure a plentiful intake of iron. In cases of iron deficiency anemia and other diseases caused by iron deficiency, iron supplementation is certainly important. However, excessive iron intake when a lack of iron is not the cause of disease is dangerous, so it is crucial to properly monitor iron levels. We reaffirmed the importance of appropriate iron intake from our knockout mouse experiments.
Restoring lysosomal function and ensuring that iron does not build up inside lysosomes would lead to the recovery of cardiac function in knockout mice. To achieve this, it is necessary to maintain NAD+ levels, so we investigated what can be done to improve NAD+ levels.
First of all, we explored what links lysosomal function to NAD+. Our work led us to one of the metabolic pathways: glycolysis. Lysosomes use a molecule called adenosine triphosphate (ATP)—which is called the energy source for biological activity—to maintain the environment required for catabolic enzymes to function. We discovered that NAD+ is closely connected to ATP production. It turned out that NAD+, which is generated in the metabolic process through which glycolytic enzymes turn sugars into energy, is required for ATP production. Consequently, if NAD+ levels fall, production of the ATP needed for lysosomal activation also declines.
We then wondered whether functions would recover if we could increase NAD+ levels. Accordingly, we provided the mice with nicotinamide mononucleotide (NMN), an NAD+ precursor (a substance generated prior to NAD+ synthesis), and examined whether cardiac function recovered. When we added NMN to the drinking water of cardiomyocyte-specific p32 conditional knockout mice to ensure they consumed it, the lifespan of the mice increased. Not only was there an improvement in the heart failure markers used to diagnose heart failure and assess its severity, but we also observed an improvement in tissue fibrosis (Figure 4 and 5). In addition, we confirmed that their previously reduced NAD+ levels had improved. As such, it would appear that there were also positive effects on lysosomal function, with an improvement in iron accumulation and a reduction in ferroptosis-induced cell death. We were able to confirm that, as a result of NMN administration, the prognosis for the dilated cardiomyopathy and heart failure stemming from mitochondrial dysfunction improved, with the mice showing a tendency toward improvement in these diseases. There is a possibility that NMN could improve secondary organelle dysfunction resulting from mitochondrial dysfunction, and also bring about a small rise in the energy supply that declines overall as a result of aging. However, even NMN administration did not lead to an improvement in the fundamental cause of disease, namely mitochondrial function itself.
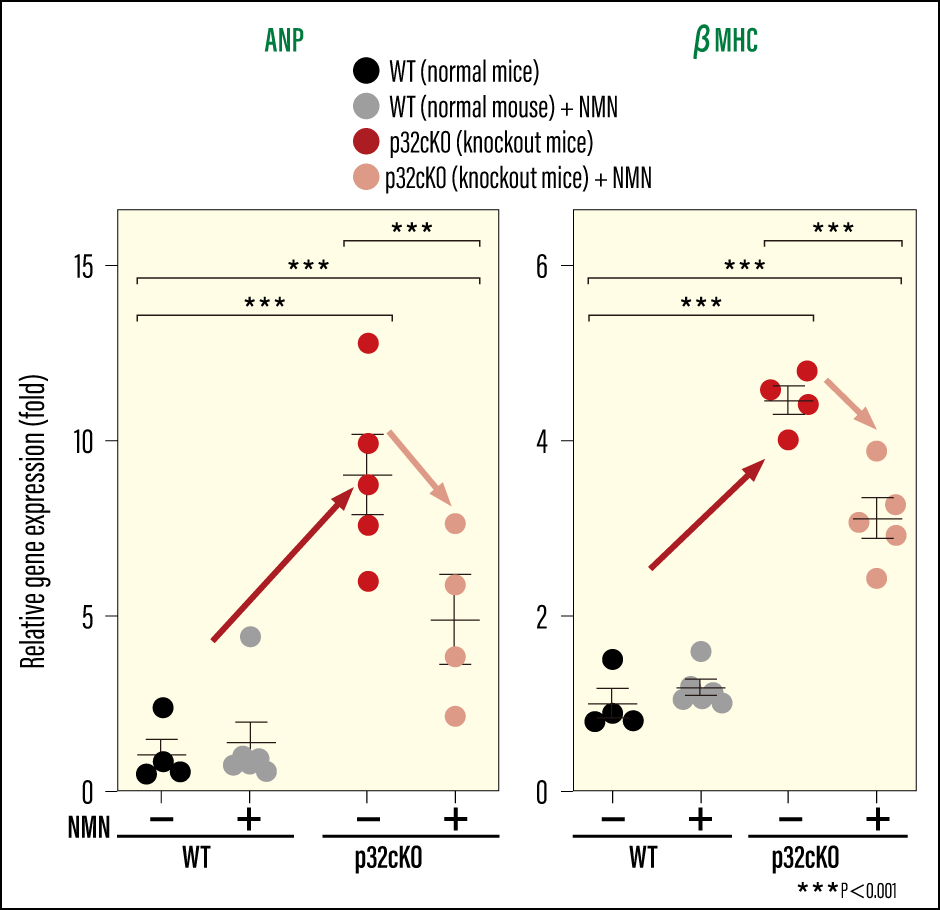 Yagi M. et al: Life Science Alliance 2023. DOI: 10.26508/lsa.202302116
Yagi M. et al: Life Science Alliance 2023. DOI: 10.26508/lsa.202302116
Figure 4. Effect of NMN administration on heart failure markersIn the knockout mice showing increased gene expression of the heart failure markers ANP and βMHC, both markers were reduced by NMN administration.
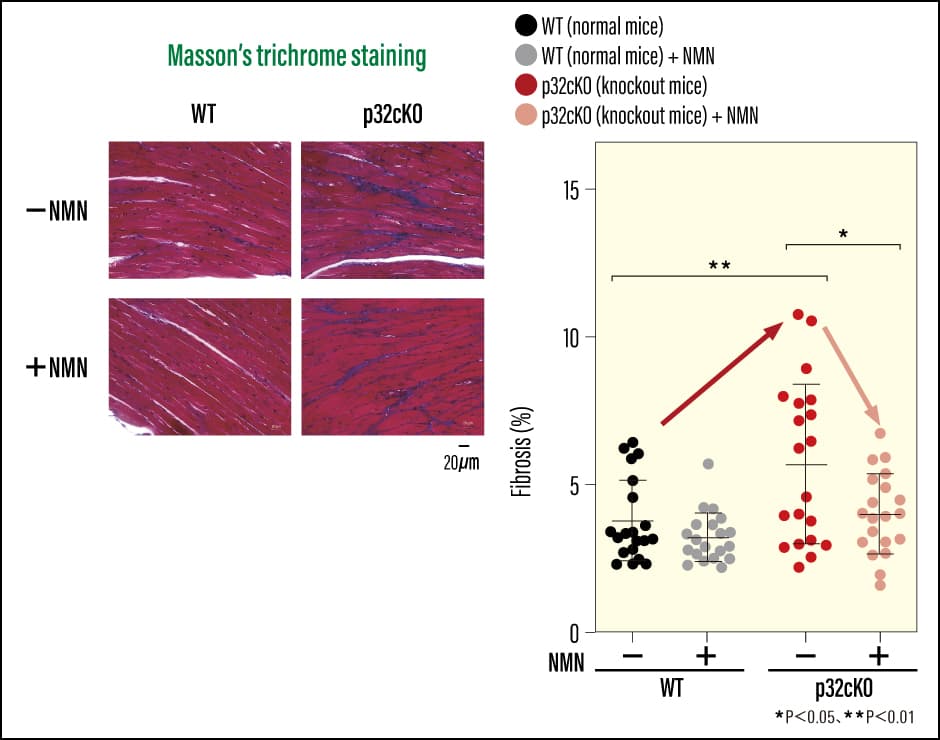 Yagi M et al: Life Science Alliance 2023. DOI: 10.26508/lsa.202302116
Yagi M et al: Life Science Alliance 2023. DOI: 10.26508/lsa.202302116
Figure 5. Effect of NMN administration on cardiac fibrosisThe research team used MT staining to check the cardiac tissue. The knockout mice had extensive fibrotic areas, as indicated by blue staining, but fibrosis was reduced following NMN administration.
The impacts of mitochondrial dysfunction
NMN is a substance derived from vitamin B3, which is produced naturally in the body. Lately, it is being sold as a supplement anticipated to have cell rejuvenation and anti-aging effects by aiding energy production and DNA repair. While our experiments focused on knockout mice, the results accorded with the theory that NMN delays the progression of aging. Foods such as edamame, broccoli, and avocado are said to contain trace amounts of NMN.
The number of mitochondria in a single cell varies from one organ to another. Furthermore, the impacts of mutations and deletions of mitochondrial DNA on cells and aging also differ from person to person. Going forward, it will be necessary to investigate how mitochondria are connected to individual organs and diseases.
For example, Parkinson’s disease is a neurodegenerative disorder in which the number of dopamine neurons in the brain decreases. We know that when mitochondrial function declines, neurite outgrowth deteriorates, and the function of cells called oligodendrocytes—which form the myelin sheath that ensheathes these nerve cell projections—also declines, making it hard for neurites to transmit information properly. In addition, PINK1 and other genes that are implicated in Parkinson’s disease are linked to mitochondrial function, and there are reports that mitochondrial dysfunction also causes autophagic dysfunction.
When it comes to aging, there is no single specific cause; rather, it is thought that mitochondrial dysfunction might have some kind of effect on other organelles, for example, thereby impairing homeostasis and leading to overall deterioration. As we see it, age-related diseases occur in response to functional decline occurring in a coordinated manner across multiple organs and organelles. Even if we cannot fully compensate for the mitochondrial dysfunction that is the root cause of disease, we might be able to remedy the secondary disorders stemming from it.
This is another reason why it is crucial to shed light on the molecular mechanisms of disease by analyzing their causes in detail. Based on the molecular mechanisms thus elucidated, individuals may be able to consume specific foods or supplements to modestly enhance their metabolism and, even if it is not feasible to cure the underlying cause of a disease, alleviate subsequent pathological changes and achieve gradual improvements. I believe this is the secret to slowing the aging process and extending healthy life expectancy.
(Figures courtesy of Mikako Yagi)