Epigenetics is a mechanism that changes the way genes are used, without changing the DNA sequence itself. The mechanism determines when and where genes are turned on or off by using DNA methylation to switch genes off, and also by finely controlling the switch by regulating the action of histones, the proteins around which DNA is wrapped. The crux of the matter is that even where genetic information is the same, gene function is influenced by environmental factors and life experiences. As such, epigenetics has become a major focus of research in many fields, including cancer, metabolic diseases, and other lifestyle diseases.
Special Feature 1 – What We Have Learned About Epigenetics So Far The two mechanisms controlling gene function
composition by Rie Iizuka
illustration by Koji Kominato

Hiroyuki Sasaki
University Professor and Professor Emeritus, Kyushu University
Completed a PhD program at Kyushu University Graduate School of Medical Sciences in 1987. In 1993, he was appointed associate professor at Kyushu University Institute of Genetic Information. In 1998, he took up the post of professor at the National Institute of Genetics. He was appointed distinguished professor at Kyushu University Medical Institute of Bioregulation in 2010, and has held his current post since 2022. The focus of his research is epigenetics and genetics, and he is keen to convey the fascination of science. His hobbies are mountain climbing and visiting hot springs.
The term epigenetics is derived from the word “epigenesis,” a neologism coined by combining the Greek prefix “epi,” meaning “after,” with the English word “genesis,” meaning “creation” or “development.” It refers to changes in gene function that occur independently of changes in the DNA sequence in the process of body development and cell differentiation, and are passed onto daughter cells following cell division. It is a mechanism that determines which genes will or will not be used in each cell, and maintains that information over a long term.
Our entire body originates from a single fertilized egg. Tissues and organs, such as liver and brain, are produced through repeated cell division starting from a fertilized egg, which differentiates into a variety of cells. Each differentiated cell has all the estimated 20,000 or so human genes, but the genes actually used vary according to the type of cell. If we look at each of the approximately 37 trillion cells constituting our body, we can see that only a proportion of the genes are actually functioning; for example, red blood cells, which are specialized in producing hemoglobin, the number of functioning genes is just 100–200 or so. It is epigenetics that enables the selective use of genes. In the case of mammals including humans, this pattern of gene usage is re-established from scratch (from the initial state in the fertilized egg) in each generation. However, there are reports suggesting that some changes in gene usage caused by environmental factors or those occurring by chance might potentially be carried over to the next generation; research on such epigenetic inheritance is ongoing.
To understand epigenetics, let us take the example of induced pluripotent stem cells (iPS cells). iPS cells are cells that have been reset to the initial state of cell differentiation (reprogrammed) by introducing a specific set of genes into a somatic cell such as a skin or blood cell. iPS cells can therefore differentiate into any kind of cell, just as cells do immediately after fertilization. In this reprogramming process, all the epigenetic patterns in the cell are reset, creating an initial state close to that of a fertilized egg capable of becoming any kind of cell.
Methylation switches genes off
For simplicity, we here classify the epigenetic mechanisms into two types. One is DNA methylation. DNA is composed of four bases: guanine (G), adenine (A), thymine (T), and cytosine (C). Methylation of DNA is the reaction that adds a methyl group (CH3) to C. To put it very succinctly, when the part of DNA that regulates gene activity is methylated, the gene is switched off, whereas a gene with a non-methylated regulatory region can be more readily switched on. DNA methylation is a simple, one-way mechanism for suppressing gene activity.
In contrast, histone modification is diverse and complex. Histones are proteins consisting of chains of amino acids, around which DNA is wrapped (Figure 1). Not only methylation, but also other types of histone modification exist, including acetylation, phosphorylation, and ubiquitination. The biological meaning varies according to which modification occurs on which amino acid and also in which combination, allowing gene switches to be adjusted across subtle gradations: “fully on,” “fully off,” “partially suppressed,” and “easily switched on.” These modifications also increase the precision of gene expression while working in harmony with or against each other.
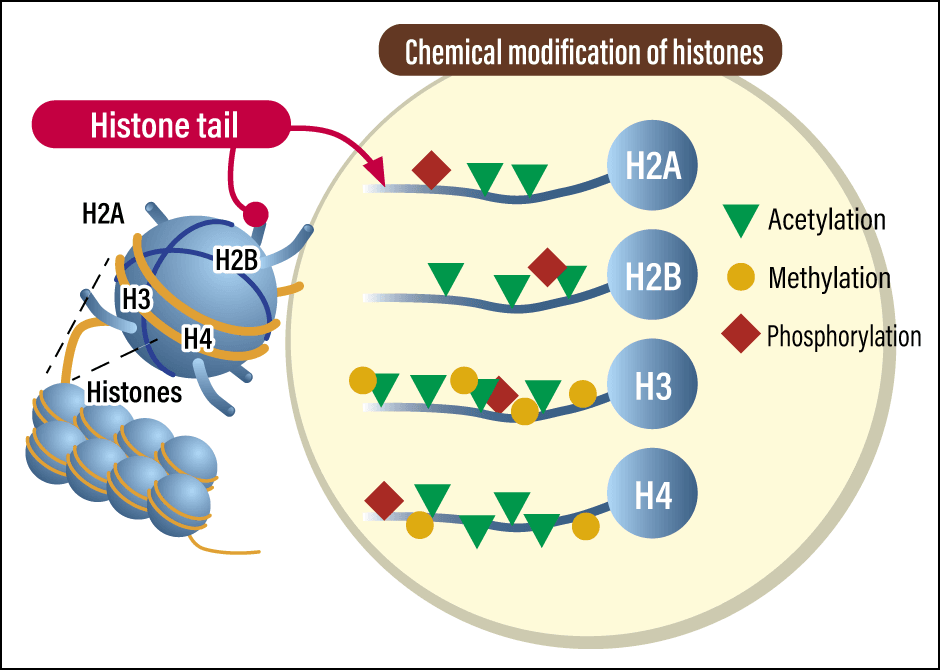
Figure 1. Epigenetic modifications of histonesA diverse array of epigenetic modifications occur in histones, which are proteins associated with DNA in cell nuclei. DNA is wrapped around a histone octamer, an assembly consisting of two molecules each of the histones H2A, H2B, H3, and H4. The chemical modifications of histones are carried out by various enzymes.
Epigenetic pattern changes during the fetal stage, starting immediately after fertilization, through cell differentiation and development, but also throughout our lives after birth, potentially influenced by several factors, including stress, environment, and nutrition. Usually, this refers to long-term control of gene expression, but brief changes in histone modification lasting for a few hours to a few days may also be caused by environmental changes or exposure to specific chemicals. For example, if DNA is damaged by ultraviolet rays, radiation, or the like, histones are marked by specific modifications indicating the location of damage to attract repair proteins. Once the repair is complete, the marker is removed and the histones return to their original unmodified state. The repair of day-to-day cell damage is also the work of epigenetics.
The reactions involving modifications are mediated by three types of agents. Writer enzymes (modifying enzymes) introduce chemical modifications, such as a methyl group or acetyl group, onto DNA or histones. Reader proteins recognize the modifications and switch genes on or off. Eraser enzymes (de-modification enzymes) remove modifications once no longer needed (Figure 2). The collaboration of these three agents contributes to cellular memory that enables cells to maintain their own identity.
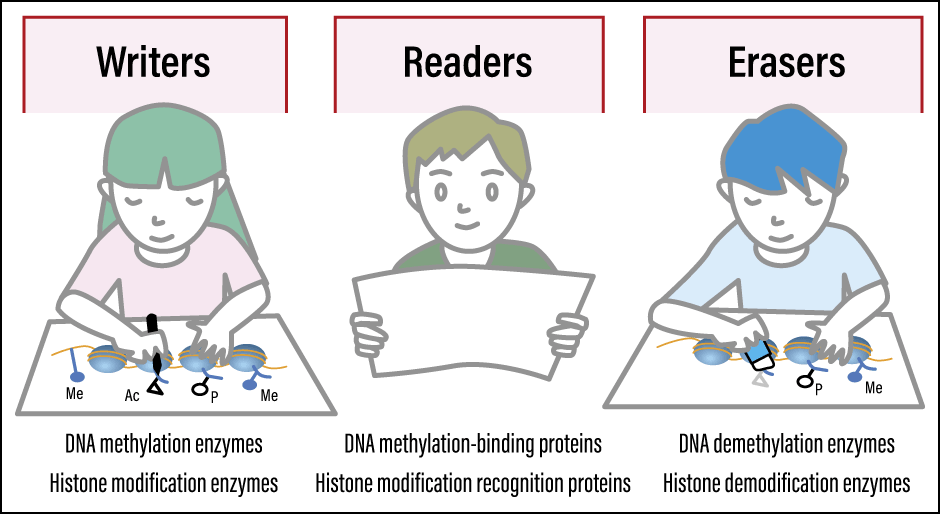
Figure 2. Basic elements of the epigenetic control mechanismThere are three key elements in the epigenetic control mechanism. DNA methylation enzymes and histone modification enzymes write modifications. DNA methylation-binding proteins and histone modification recognition proteins read modifications and switch genes on or off. DNA demethylation enzymes and histone de-modification enzymes erase modifications that are no longer required.
Risk of gastric cancer may remain even after eradication
Recent research has revealed that epigenetic abnormalities are involved in the mechanisms behind an assortment of diseases. There is a particularly strong association with cancer. For example, Helicobacter pylori (H. pylori) infection is regarded as the leading cause of gastric cancer; the infection results in chronic inflammation of the gastric mucosa, causing changes in the DNA methylation pattern. Scientists have also discovered cases in which methylation does not return to its original pattern even once H. pylori has been eradicated, leaving the patient at a high risk of developing gastric cancer, and the degree of methylation is being applied to predicting the onset of the disease.
A number of epigenetic drugs have been developed. For example, when a cell becomes cancerous, changes may occur, such as an increase in methylation (suppression) of genes that control cell division or repair mutations; therapeutic drugs that control this process have been developed. Drugs that inhibit DNA methylation enzymes are effective in some patients with myelodysplastic syndromes (MDS; a condition sometimes described as pre-leukemia), for which there were hitherto hardly any effective drugs.
Drugs that target histone modification have also appeared on the scene. Histone deacetylases (HDACs) are enzymes that remove acetyl groups from histones and inhibit transcription; in cancer cells, HDACs may be overactivated, silencing tumor suppressor genes. HDAC inhibitors, which block this phenomenon, have been approved for use against a number of blood cancers.
However, crucial issues still remain. DNA methylation enzymes and HDACs are enzymes that affect the function of many genes in the body. That means they act on not only target genes, but also other genes, and so side effects have been reported. There are also aspects where the mechanism of action has not been fully clarified; scientists know that they work, but do not yet have a clear picture of the reasons why. Epigenetic drug discovery also aims at a range of other diseases, but the development of technology for selectively targeting genes of interest is a major challenge.
The relationship between epigenetic abnormalities and diseases of the brain and nervous system is also attracting attention. There have been reports on patients with Alzheimer’s disease having reduced methylation levels in regulatory regions of specific genes. Accumulated data also suggests that epigenetic changes in the glial cells that support neurons are related to the progression of dementia and neurodegenerative disorders. Studies that reframe disorders of the brain from an epigenetic perspective are expanding rapidly.
In addition, regarding autism spectrum disorder (ASD), it has been reported that numerous mutations are found around genes that regulate the association of histones with DNA, and their functions. Furthermore, while DNA methylation normally works to suppress DNA sequences called transposons, scientists have pointed out derepression of transposons in ASD. Based on indirect evidence of this kind, many researchers believe that epigenetic abnormalities are, in some way, involved in the etiology of ASD.
Regarding the relationship between lifestyle diseases and epigenetics, there is a famous study focused on a historical event.
Toward the end of World War II, a Nazi Germany-imposed blockade on food supplies caused a severe famine in the western Netherlands. It became apparent that children born to mothers who were pregnant during this period frequently went on to develop diseases including obesity, hypertension, ischemic heart disease, and type 2 diabetes mellitus in adulthood. Scientists believe that epigenetic modification patterns altered as a result of their lacking adequate nutrition at the fetal stage, causing them to develop an energy-conserving metabolic profile, which persisted after birth. People who experienced famine while in the womb are also reported to have had reduced DNA methylation of the insulin-like growth factor 2 (IGF2) gene.
As well as being crucial to development and cell differentiation, epigenetics is also responsible for etching the impacts of lifestyle and environment onto the body. One of the best illustrations is the epigenetic clock, which is a major topic of discussion right now. Derived from the correlation between DNA methylation patterns at various locations on the genome and a person’s actual age and health condition, this tool makes it possible to estimate a person’s biological age. One could say that it visualizes the marks left on the genome (and eventually the body) by disease and lifestyle. For example, in patients who are positive for human immunodeficiency virus (HIV), the biological age estimated by the epigenetic clock tends to shift toward a older state than their actual age; in other words, that they appear to be aging faster.
Private sector services that use the epigenetic clock in lifestyle-related consultations have emerged, while DNA specimens left at crime scenes are being used to estimate the age of individuals connected to the case. Consideration is reportedly given to the application of the epigenetic clock by life insurance companies. Even though it remains unclear what individual methylation patterns mean in biological terms, we have obtained a useful yardstick with the aid of machine learning.
Attention has come to focus on the potential of epigenetic modifications as biomarkers, as typified by the epigenetic clock. Epigenetic patterns differ according to cell type, with each of the more than 200 human cell types—several thousand types, if you divide them into finer categories—having its own epigenome. The International Human Epigenome Consortium (IHEC), to which Japan contributes, was established to decode them. By putting in place “normal” reference data, it will become easier to develop technologies for identifying abnormal epigenetic patterns in patients’ cells.
While many differences in our characteristics and individual features are down to differences in the DNA forming the base of our makeup, it is not only our genes that determine our biological functions; epigenetics also has a major impact.
The mechanism behind the three colors of a calico cat’s fur
A familiar example of epigenetics can be found in calico cats (Figure 3). Calico cats have tricolored fur in white, orange, and black, and this is related to an epigenetic phenomenon called X chromosome inactivation in females (calico cats are nearly always female).
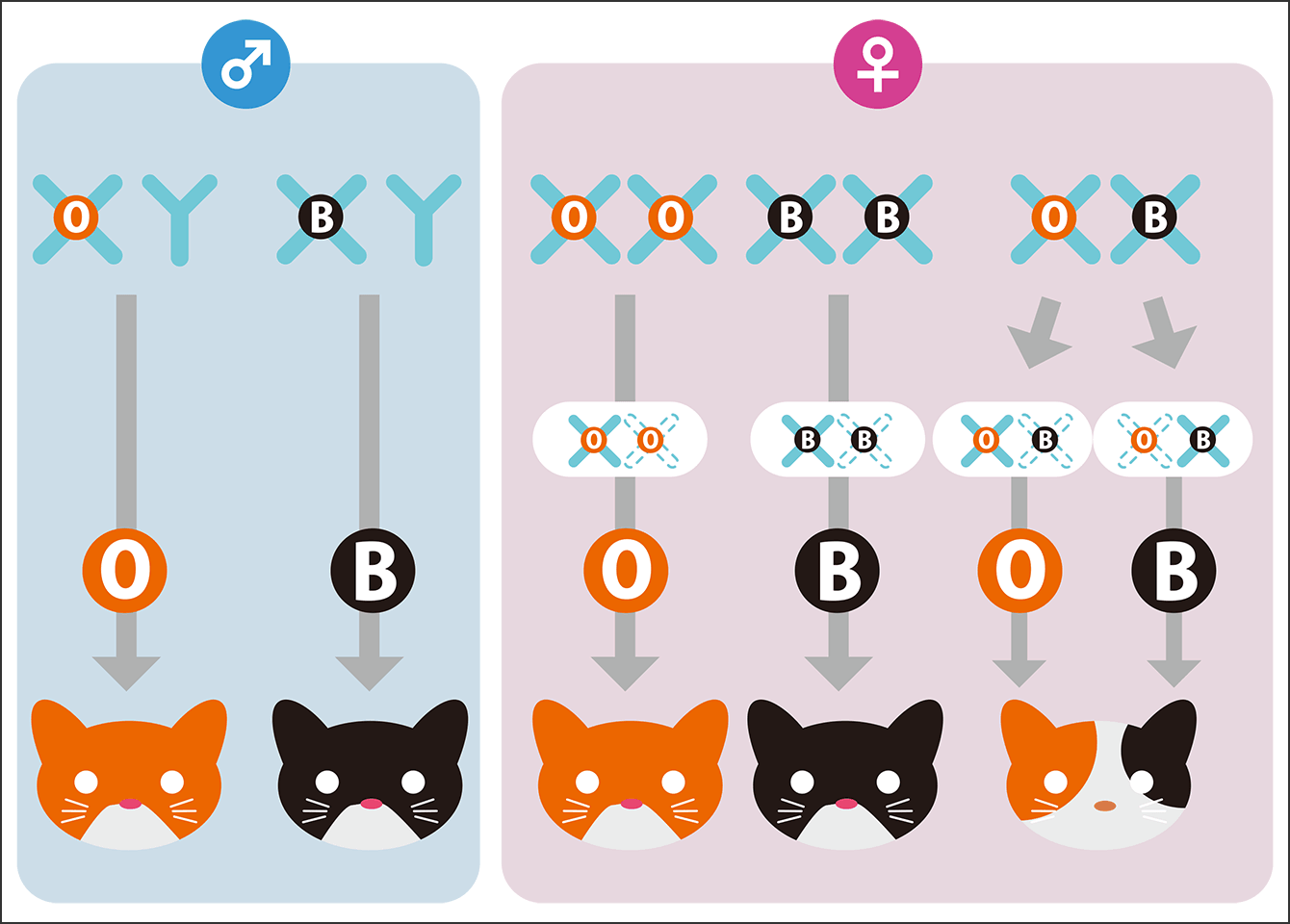
Figure 3. The mechanism determining the patterns on calico catsThe black and orange patches in a calico cat’s tricolored fur are caused by X chromosome inactivation involving epigenetic modifications. As the genes determining whether the fur will be black or orange are located on the X chromosome, only one of these colors appears in males, which have only one X chromosome. On the other hand, because females have two X chromosomes, they can possess the information for both orange and black, forming the basis of a calico cat’s fur. During early development, one of the two X chromosomes is chosen at random to be inactivated by means of epigenetic modification in each cell. Cells that express the orange gene produce orange pigment, while cells that express the black gene produce black pigment. Pigment cells then spread out across the surface of the body and occupy certain regions, forming different tricolor patterns in each individual. As the white color is due to the action of a different gene, it is unrelated to X chromosome inactivation.
Of the genes that determine fur color, the gene responsible for white fur is located on an autosome, which is a chromosome present equally in male and female. On the other hand, the gene that switches the production of black (eumelanin) or orange (pheomelanin) pigment is located on X chromosome. As males have only one X chromosome, only one color will emerge, either orange or black. In addition to calico cats, tortoiseshell cats, which have bicolored fur in black and orange, are virtually all female for the same reason.
Although females have two X chromosomes, it is deleterious to have both of them active, so one of the X chromosomes is randomly chosen for inactivation at an early stage during development. Some areas become white due to the action of the gene located on an autosome, while the rest will become either orange or black, depending on which X chromosome is inactivated, thereby creating a tricolored pattern. In other words, the color of the pigmented parts of a calico cat’s fur is determined by chance through an epigenetic mechanism called random X chromosome inactivation. This is precisely why even genetically identical cloned cats will not have exactly the same tricolored fur pattern.
However, from the time the hypothesis of calico cat patterns was propounded in 1961 right through until the present post-genome era, the gene on the X chromosome had, for some reason, not been identified. Accordingly, I set out to identify the gene, with the cooperation of those around me.
The domestic cat (Felis catus) diverged from the African wildcat (Felis lybica) around 10,000 years ago. As the African wildcat is dark brown in color, bright orange is a newly emerged mutation. It occurred to me that in that case, we might be able to identify the gene if we looked for a mutation on the X chromosome found only in cats having orange fur. When we analyzed the genomic DNA of cats with various fur colors that we had collected in Fukuoka and compared their DNA sequences, we discovered a deletion of around 5,000 bases within a gene called ARHGAP36 on the X chromosome of cats with orange fur. Furthermore, when we compared our findings with the cat genome data available from the University of Missouri in the U.S., the presence or absence of the deletion coincided exactly with whether or not the cats had orange fur.
We then investigated the deleted region further and found that it was a DNA sequence called an ultraconserved element found across many animal species, including humans, mice, dogs, and chickens. Scientists believe that ultraconserved elements control gene expression; as calico cats lack this sequence, expression of the ARHGAP36 gene is dysregulated and the ARHGAP36 protein is overproduced. A larger amount of ARHGAP36 protein suppresses eumelanin synthesis, resulting in synthesis of more pheomelanin that gives orange color. Thus, after 60 years, we have shed light on the mechanism responsible for calico cats’ orange fur.
(Figures courtesy of Hiroyuki Sasaki)