Among the many diseases caused by gene mutations, Duchenne muscular dystrophy (DMD) is an intractable disease associated with severe outcomes. The patient’s body is unable to synthesize a protein called dystrophin, which leads to the loss of muscle tissue. With few options available to cure the disease, hopes are currently pinned on a combination of genome editing to correct the causative gene mutation, therapy to restore normal protein synthesis, and transplantions of cells derived from induced pluripotent stem cells (iPS cells).
Special Feature 1 – An Introduction to Genome Editing Gene editing and iPS cell therapy offer hope for muscular dystrophy patients
composition by Takakazu Kawasaki

Akitsu Hotta
Principal Investigator (Junior Associate Professor ), Department of Clinical Application, Center for iPS Cell Research and Application (CiRA), Kyoto University
Graduated from the Department of Chemical and Biological Engineering at Nagoya University’s School of Engineering in 2001. In 2006, he completed the doctoral program in biotechnology at Nagoya University’s Graduate School of Engineering, earning a Ph.D. in bioengineering. The same year, he took up a post as a postdoctoral research fellow in the Developmental & Stem Cell Biology program at the Hospital for Sick Children in Toronto, Canada. At Kyoto University, after serving as a principal investigator (assistant professor) at the Institute for Integrated Cell-Material Sciences (iCeMS) and in the Department of Reprogramming Science at the Center for iPS Cell Research and Application (CiRA) in 2010, he was promoted to junior associate professor in the Department of Life Science Frontiers at CiRA in 2016. He took up his current position in 2018.
About 330 diseases have been designated by the Japanese government as intractable/rare diseases, and around half of them are known to be caused by or associated with gene mutations. Diseases stemming from gene mutations are hard to treat and are congenital, meaning symptoms of the disease appear from childhood.
Muscular dystrophy is one of the diseases arising from gene mutations. There are a number of different forms of the disease, including myotonic dystrophy and Becker muscular dystrophy, but the most serious and most common type is Duchenne muscular dystrophy. The number of children suffering from muscular dystrophy in Japan is estimated at somewhere between 2,500 and 4,500. Because the causative gene’s mutation is found on the X chromosome of the sex chromosomes, the majority of patients are male.
Every movement destroys muscle tissue
Most mammals have sex-determining chromosomes, with males having XY chromosomes and females XX. Most muscular dystrophy patients tend to be male, because females (XX) have two copies of the dystrophin gene, so even if one copy is mutated in females, the other gene can ensure normal dystrophin protein synthesis.
In healthy individuals, increased muscle movement has a weight training effect, causing the muscles to become stronger and thicker. However, a muscular dystrophy patient’s muscles lack the important structural protein called dystrophin found on the inside surface of the muscle membrane. As a result, the more the patient’s muscle moves, the more muscle tissue is destroyed. Inevitably, atrophy of the muscle tissue progresses, and by their teens, patients have to use a wheelchair in daily life, as they are unable to walk or stand unaided, and they become more or less bedridden by their 20s.
DMD is named after the French neurologist Guillaume B.A. Duchenne, who described the disease in the 1860s. Subsequent progress in molecular biology resulted in the 1986 discovery by Dr. Louis M. Kunkel of Boston Children’s Hospital in the U.S. that the disease was caused by the defect of the dystrophin gene.
All organisms on earth have nucleic acids called deoxyribonucleic acid (DNA) and ribonucleic acid (RNA). These nucleic acids contain the entire blueprint that shapes each organism. Nucleic acids consist of a set of three bases from among the following four types: adenine (A), thymine (T), guanine (G), and cytosine (C). The sets specify which amino acids should be used to synthesize proteins, issuing instructions like “commence the protein synthesis” and “terminate the protein synthesis.”
The dystrophin gene itself is huge, consisting of 2.2 million bases. However, if just a few of these bases within the gene are compromised, the body fails to synthesize the dystrophin protein. For example, if part of the gene that produces dystrophin mutates such that the base sequence AAA becomes TAA, the instruction terminates the protein synthesis prematurely and fails to synthesize functional dystrophin protein. It is theorized that by genome editing the mutation to the correct sequence, the body will be able to synthesize dystrophin appropriately.
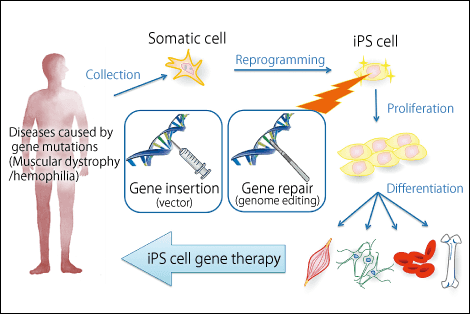
Figure 1. A new gene-therapy strategy using iPS cellsResearchers aim to develop safer human iPS cell production and selection methods in an effort to create new gene therapies for hemophilia and other congenital diseases.
Therapeutic effects maintained long-term
The reason why I started studying treatments based on gene editing is related to the fact that I had poor health in my childhood. From my elementary school days, I often missed school, because I suffered from asthma. I struggled to breathe when I had an asthma attack, but as soon as I took steroids and other drugs, the symptoms vanished and I was able to breathe again. While that made me marvel at the power of medicine, the drugs would stop working as soon as they were excreted from my body.
Although I was interested in pharmaceuticals, I did not have a strong desire to become a medical student. While medicine is a noble profession, in which physicians try to help the patients who come to see them, I want to be of use to society as a whole through my research output. Plus, I am not keen on the sight of blood even now.
Since drugs are made up of chemical compounds, I entered the chemical engineering program at Nagoya University’s School of Engineering. While I was there, I happened to encounter genetic engineering via the biological engineering program and I realized that if you could edit genes, you could control various attributes in organisms, and also that, in the case of people suffering from diseases caused by gene mutations, curing the gene mutation would ensure that the patients stayed cured for the rest of their lives. This is why I entered the field of genetic engineering research.
Until I got my degree, I was involved in research focused on introducing genes into chicken’s eggs, but I wanted to undertake research in embryonic stem cells (ES cells), so I decided to go and study at the Hospital for Sick Children in Toronto. Toronto’s Hospital for Sick Children is North America’s largest children’s hospital and, almost like a department store, it has all kinds of systems in place to enable children suffering from various illnesses to keep their spirits up. There were many pediatric inpatients suffering from muscular dystrophy or other diseases caused by gene mutations. I was involved in the development of viral vectors, which are used in gene therapies to carry therapeutic genes into the bodies of patients suffering from sickle-cell anemia or hemophilia.
In 2006, while I was conducting research in Canada, I learned about Dr. Shinya Yamanaka’s article on induced pluripotent stem cells (iPS cells). Initially, I could not believe that it was true, but when I tried it for myself, I could immediately generate iPS cells, just as the article had said. I was astonished and dove right into iPS cell research.
Genome editing holds out the possibility of a cure
It was 2010 when I joined the Center for iPS Cell Research and Application (CiRA) at Kyoto University. Dr. Yamanaka accepted me, and when he handed over my official letter of appointment certifying me as a CiRA researcher, he told me, “Here at CiRA, I want you to work on developing technologies that will make it possible to use iPS cells to transcribe a single base in a genome (the whole set of genetic information in a body).” I was aware that genome editing technology would be needed to make use of iPS cells, so I shifted to genome editing technology research.

(Left) Dr. Shinya Yamanaka in front of the Center for iPS Cell Research and Application.
(Photograph courtesy of the Center for iPS Cell Research and Application, Kyoto University)
(Right) Human iPS cells. A colony of human iPS cells established from fibroblasts. The width of the colony is about 0.5 mm.
(Photograph courtesy of Dr. Shinya Yamanaka, Kyoto University)
There are two reasons why I chose DMD as the subject of my research for genome editing therapies at CiRA.
The first is that DMD is, as stated above, the most serious and the most common form of muscular dystrophies found in children. The conventional gene therapy is gene supplementation therapy, which involves inserting a whole, correctly functioning gene into the cell from outside to complement the broken, non-functioning gene. However, because the dystrophin gene, which causes muscular dystrophy, is huge—one of the top 3 largest in the human genome by size—it has been difficult to treat muscular dystrophy using conventional gene supplementation therapy. It occurred to me that genome editing, which enables gene mutations to be repaired directly, held the possibility of a cure.
The second reason is that I was fortunate enough to meet Dr. Hidetoshi Sakurai (associate professor), who was appointed to CiRA at the same time as me. Dr. Sakurai conducts research on the differentiation of iPS cells to skeletal muscle stem cells and then using these cells to replicate the pathology of muscular dystrophy and other diseases and to search for therapeutic drugs. We have the chance to discuss various topics every day because we shared the same office, and in the course of learning about muscular dystrophy from him, I realized that it might be possible to use genome editing to tackle this disease.
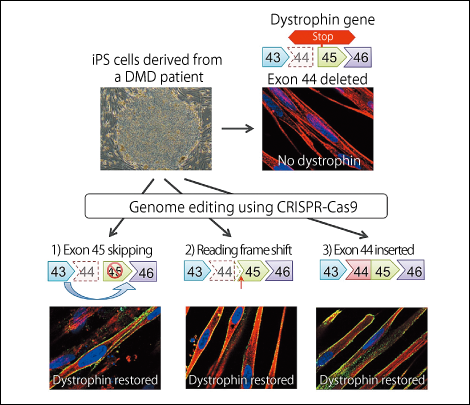
Figure 2. Technique for repairing the gene mutation responsible for Duchenne muscular dystrophy (DMD)Three different methods were used to carry out the gene editing of exon 44 with CRISPR-Cas9 in cells from a DMD patient, and all three were successful.
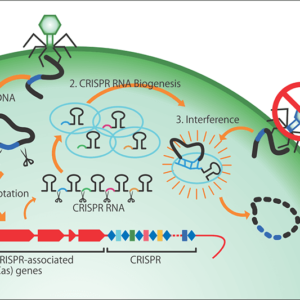
At the time, the only technology available for editing genomic sequences in a cell uses enzymes called zinc finger nucleases (ZFNs), and it cost about 3 million yen to make just one set of ZFNs. Then, in 2012, a novel technology was developed that used an enzyme called CRISPR-Cas9, and the price of reagents for research purposes came down tens of thousands of yen, making this technology more accessible than ZFNs. To edit genes, researchers create a short piece of RNA that serves as an instruction for which part of the gene to cut; this too can be done in the laboratory for just a few thousand yen.
Gene editing using CRISPR-Cas9 is a task that involves painstakingly searching a gene’s lengthy character strings—a bit like an encyclopedia—to find the target location, then cut it out and repair it. CRISPR-Cas9 can find the target portion of a huge gene and snip it within a few hours. After that, the cell itself takes care of the repair of the gene, which is accomplished within no more than several minutes to a few dozen minutes. The genomes in our bodies can be cut by ultraviolet rays or radiation every minute, which could cause cell death or cancerous changes if left unchecked, so our cells are equipped with a self-repair mechanism.
We previously succeeded in producing iPS cells from a patient with DMD, in which the dystrophin protein reading frame is disrupted, by missing the dystrophin gene’s 44th exon (the part carrying information about protein synthesis). We devised three strategies for repairing this mutated dystrophin gene.
1) Rendering the 45th exon illegible and joining the 43rd exon to the 46th to restore the reading frame.
2) Inserting one base to restore the reading frame.
3) Inserting the 44th exon to restore the full length dystrophin proten.
When we differentiated iPS cells repaired using these methods into skeletal muscle cells, we found that all of the repair methods resulted in the production of dystrophin protein.
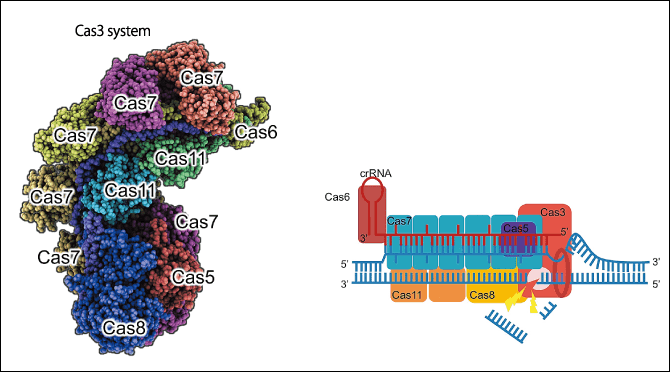
Figure 3. A new genome editing tool, CRISPR-Cas3Because CRISPR-Cas9 and other genome editing tools can only induce cuts of short lengths and there are problems with intellectual property rights, Dr. Hotta and his team have developed a new genome editing tool.
Safer than using a virus
Recently, I have been working on research involving nanocapsules for delivering CRISPR-Cas9 to a patient’s muscle tissue. Normally a virus is used as a vector (delivery agent), but then the virus itself remains in the cell and continues to express the enzyme for years. If CRISPR-Cas9 DNA cutting enzyme continues to act inside the cell, there is a growing risk that the Cas9 might cut genes other than the site that it was meant to snip. Accordingly, if we can use a means other than a virus to transiently deliver the CRISPR-Cas9 enzyme to muscle tissue, it will be much safer than using a virus. It is hoped that this method of repairing genome mutations in the cells inside a patient’s body will prevent further destruction of their muscle tissue and that rehabilitation will enable them to maintain and use their muscles once cured.
However, once muscular dystrophy progresses to its later stages, muscle fibers are destroyed and replaced by fibrous and adipose tissues, so the muscle will not be restored by repairing the genomic gene. If skeletal muscle cells and muscle stem cells are produced from iPS cells and transplanted into the body, there is a possibility that they might be able to produce new muscle fibers. Dr. Sakurai is researching this therapy.
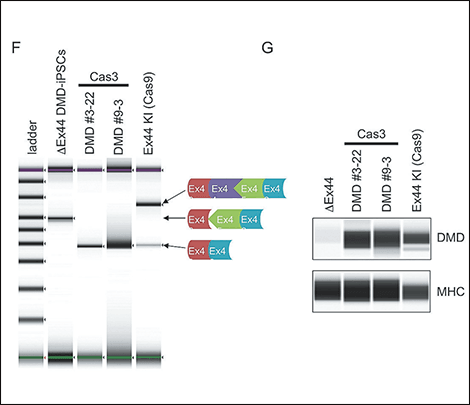
Figure 4. Exon-skipping therapy using CRISPR-Cas3CRISPR-Cas3’s cutting pattern was analyzed, and dystrophin protein was successfully restored by introducing a deletion further up the target sequence and thereby eliminating the mutated portion.
In summary, our gene editing therapy is effective before the muscle fiber cells are destroyed, but we believe that the latter cell transplant therapy will be crucial if the muscle fibers have already been destroyed. They are both important therapies, so research into both medical technologies is required.
Because genome editing is a new technology, we must test its safety and effectiveness very carefully.
Once gene editing technology becomes established, we should be able to apply it to other genetic diseases. Keenly aware of the earnest hopes of those who suffer from these diseases, we are continuing to pursue our research diligently.
(Figures courtesy of Akitsu Hotta)