Produced by infected tissue, chemokines induce the migration of immune cells in the blood to the site of the infection, where the cells engulf the bacteria (a process called phagocytosis), bringing down the inflammation. However, if they continue to migrate, they can also attack healthy organs, triggering rheumatoid arthritis or other autoimmune diseases. Scientists had identified that the migration of immune cells due to chemokines was involved in autoimmune disease, but the details remained a mystery. However, the development of a new imaging system capable of visualizing immune cell and chemotactic factor dynamics and analyzing them in real time has brought to light the mechanisms involved.
Special Feature 1 – The Evolution in Visualization A new imaging system visualizing the pathogenesis of arthritis
composition by Takeaki Kikuchi

Yoshishige Miyabe
Professor and Chair, Department of Immunology and Medicine, St. Marianna University School of Medicine
Graduated from Nagoya City University Medical School in 2005. He became a senior resident in the Department of Rheumatology at Tokyo Medical and Dental University's Faculty of Medicine in 2007 and was appointed assistant professor in the department in 2012. In 2013, he successfully completed the doctoral program at Tokyo Medical and Dental University's Graduate School of Medical and Dental Sciences. That same year, he took up a post in the U.S. as a postdoctoral fellow in medicine at Massachusetts General Hospital, Harvard Medical School, Boston. In 2015, he became a senior research fellow at the same institution and was then appointed an instructor in medicine there in 2017. In 2018, he took up the post of Associate Professor of Cell Biology at Nippon Medical School's Institute for Advanced Medical Sciences. He has held his current post since 2022.
All the cells from which our body is made are moving. We can live a healthy life because of the ability of these cells to move to the right place at the right time. In other words, cell migration is crucial to maintaining biological activity.
Chemokines serve as markers where immune cells assemble
Cell migration plays a very important role in host defense. For example, when a bacterial infection of skin occurs, the tissue is damaged and inflammatory mediators are released. (1) These inflammatory mediators cause immune cells circulating in the blood to migrate to the infected tissue. (2) Once they enter the tissue, the immune cells activate and can remove the bacteria by consuming them. Allow me to explain the migratory process referred to in (1). The inflamed area mainly produces chemotactic factors called chemokines, which induce cell migration (Figure 1). The circulating immune cells arrive at the marker provided by the chemokines expressed in the infected tissue. You can imagine it as something like a car on an expressway arriving at a marker in the form of a sign for a parking area. In this regulatory mechanism of cell migration, the immune cells are initially floating around in the blood as it circulates, but eventually start rolling along the vascular walls. As they roll along the vascular walls covered in chemokines and other chemotactic factors, they slow down and finally come to a complete halt. The immune cells subsequently change shape from circular to amoeboid and then crawl through the blood vessels, searching for an entrance into the infected tissue, and eventually penetrating it.
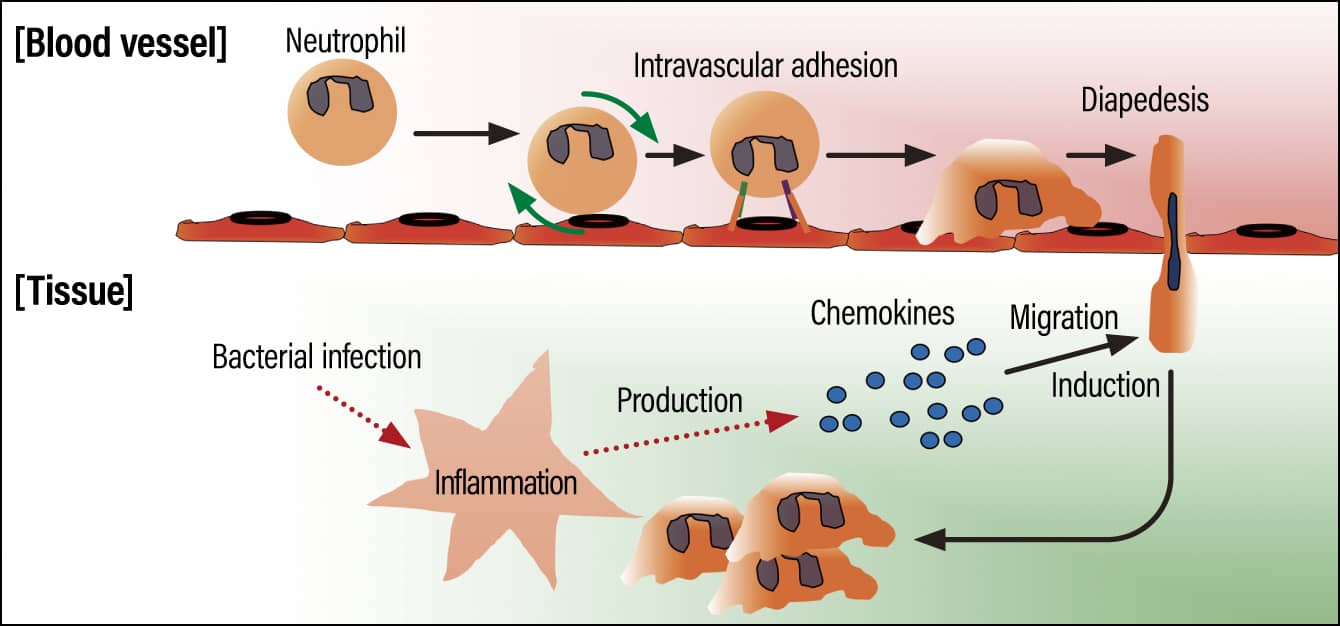
Figure 1. The regulatory mechanism of cell migrationThe immune cells that had been floating around inside the blood vessel begin to roll along the vascular wall, eventually coming to a complete halt when they reach the marker provided by the chemokines. They then transmigrate through the vascular wall (diapedesis).
Normally, once the inflammation clears up, the immune cells stop migrating. However, continual abnormal migration of immune cells can cause autoimmune diseases or other chronic inflammation (Figure 2).
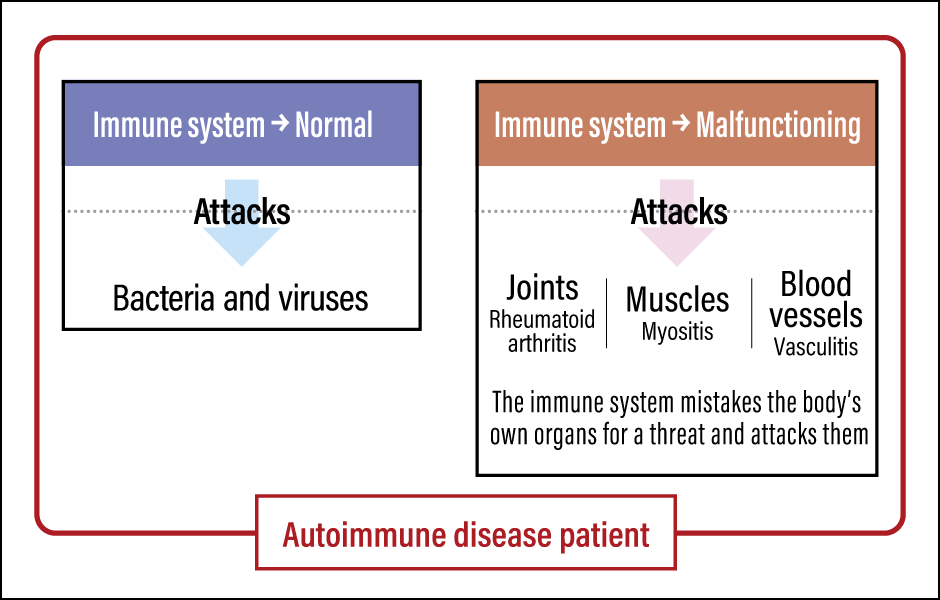
Figure 2. The mechanism of autoimmune diseasePatients with an autoimmune disease have both a normal immune system and a malfunctioning one, which repeatedly mistakes the individual’s own organs for a threat and attacks them.
An autoimmune disease is a condition in which an individual’s immune cells assume that the organs they are supposed to protect —— joints, muscles, and blood vessels —— are a threat and continually attack them. Repeated attacks on joints cause rheumatoid arthritis, those on muscles trigger myositis, and those on blood vessels give rise to vasculitis.
The approach taken in conventional immunosuppressive therapy is to suppress the activation of the immune system. This is based on the idea that the presence of immune cells is fine as long as they do not attack joint tissue, for example. Consequently, immunosuppressants were administered to patients with autoimmune diseases.
Five receptors involved in migration
In recent years, the emergence of biologic drugs has saved many patients with rheumatoid arthritis from the agony of this disease. However, they are effective in only around 30% of patients, and drug resistance and relapses remain a problem. In addition, these treatments weaken patients’ defense against disease, because they suppress the patient’s immune system, giving rise to the possibility of catching infectious diseases or other complications.
A new approach to immunosuppressive therapy was therefore developed, in the form of a treatment that inhibits immune cell migration. This works via the tissue-specific inhibition of immune cell migration to the affected tissue.
The mechanism via which immune cells enter tissues differs from one organ to another. Accordingly, if we can shed light on the mechanism regulating immune cell migration in each organ, we will be able to regulate cell migration at will in an organ-specific way. In other words, it will be possible to suppress only the immune system in the affected area, while maintaining the immune system in other healthy organs.
My research focuses on the immune cell migration mechanism involved in rheumatoid arthritis. So far, we have discovered that there are five receptors involved in the migration of neutrophils to the joints: BLT1, a lipid mediator receptor expressed in neutrophils; the chemokine receptors CCR1 and CXCR2; the complement receptor C5aR1; and the endothelial cell complement receptor C5aR2.
At the same time, we had no idea why these five receptors are needed for neutrophils to migrate to the joints. We were proceeding with our research based on the inferences we had made from the data but were frustrated by the fact that we could not look inside the living body to see whether our deductions were correct. Although we had conducted experiments using a device that simulated living organisms, it is completely different from the environment inside our bodies. In other words, there were at that time no experimental systems that analyzed cell migration in vivo.
When I was in my fourth year at graduate school, I read an article that Dr. Thomas T. Murooka (now an associate professor at the University of Manitoba in Canada) had published in the scientific journal Nature in 2012, and it had a great impact on me. Dr. Murooka was conducting HIV research using humanized mice at the time. Scientists already knew that HIV infected T cells but did not understand specifically how the infection occurred. Dr. Murooka’s article contained video images showing how HIV-infected T cells in the lymph nodes became elongated, touched uninfected T cells, and infected them with HIV. The research team were able to use in vivo imaging to see in real time what was happening inside the living organism, how the cells moved, and how one cell communicated with another. It was this that made me want to study in vivo imaging systems someday. However, it was a research technique with which few in Japan were familiar back then.
Observing neutrophils infiltrating joints
After finishing graduate school, I went to study in the U.S. at Harvard Medical School’s Massachusetts General Hospital, where I conducted research under pioneering chemokine researcher Dr. Andrew D. Luster. As luck would have it, the department was also home to Dr. Thorsten R. Mempel, a pioneer of in vivo imaging systems. Dr. Luster was originally engaged in joint research into in vivo imaging systems with Dr. Mempel. And in fact, the findings by Dr. Murooka that I mentioned above were the result of joint research he had conducted with Dr. Luster and Dr. Mempel. Dr. Luster’s research department was equipped with a two-photon excitation microscope and analysis software, which were ideal for in vivo imaging.
Thinking that it must be fate, I talked to Dr. Luster about building a system for in vivo imaging of joints. However, he was unenthusiastic to start with, telling me that eight postdoctoral fellows in his lab had already taken on the challenge of building such a system, but not one of them had succeeded. Nevertheless, I did not give up. Having managed to persuade Dr. Luster, I began working on building a system for in vivo imaging of joints as part of a collaborative research project with Dr. Mempel.
As Dr. Murooka himself was a postdoctoral fellow in Dr. Mempel’s laboratory at the time, I learned basic bioimaging techniques from Dr. Murooka and created my in vivo imaging system for joints via a process of trial and error. As a result, I succeeded in using a microscope to observe the movement of cells and molecules marked with fluorescent labels, after first shaving the lower legs of arthritic mice, carefully peeling away the skin to expose the synovial tissue, and shining a laser on the tissue. The reasons why previous attempts to create an in vivo imaging system for joints had failed were a lack of understanding about which area should be observed and the fact that the surgical procedure caused immune cells to migrate to the tissue. My own experience as a physician working in the field of rheumatology and connective tissue diseases proved useful in determining the area to observe. In the case of the surgical procedure, while conducting endless rounds of mouse surgery, I learned the knack of eliminating external factors. Six months after I began my studies in the U.S., I had completed my in vivo joint imaging system. Even now, I clearly remember the reactions of Dr. Luster, Dr. Mempel, and the other laboratory members when they viewed video images of the pathogenesis of arthritis for the first time at a lab meeting.
Using the in vivo joint imaging system I had built in this way, I set about unraveling the mechanism that causes rheumatoid arthritis.
First, I looked at the movement of immune cells. Four of the receptors expressed in neutrophils mentioned above are key to neutrophil migration in arthritis. So, how do these four receptors actually function? In an effort to find out, we induced immune complex-induced arthritis in wild type mice and in mice in which we had knocked out the genes for the respective receptors. We then undertook comparative observation to ascertain how these receptors acted on neutrophil migration in arthritic joints.
In the wild type mice, we were able to observe a very large number of neutrophils infiltrating the joint.
In the mice in which the relevant receptor genes had been knocked out, on the other hand, we observed completely different movements. We discovered that the four receptors have different roles. We found that C5aR1 and BLT1 are involved in the onset of arthritis, with C5aR1 essential to the intravascular adhesion of neutrophils, which is the starting point for inflammation, while BLT1 acts on the infiltration of the joint tissue by the neutrophils adhering to the blood vessel. At the same time, it became apparent that CCR1 and CXCR2 come into play after the onset of arthritis, with CCR1 involved in the intravascular adhesion of neutrophils and CXCR2 in the diapedesis of the neutrophils.
Incidentally, rheumatoid arthritis is classed as a type III hypersensitivity reaction mechanism. Type III hypersensitivity reaction mechanisms were originally thought to stem from the inflammation of macrophages and mast cells present in tissue. However, the joint cavity is an unusual environment, in that it is sealed off from the bloodstream and therefore has no immune cells in the form of macrophages and mast cells. The conventional type III hypersensitivity reaction mechanism could not explain the mechanism causing inflammation in the joint cavity.
With the aid of the in vivo joint imaging system, our research team discovered that the complement C5a produced in joint tissue is transported into the vascular lumen and deposited there, whereupon, mediated by the circulating neutrophil complement receptor C5aR1, it induces the intravascular adhesion of immune cells that forms the starting point for inflammation. Visualizing the pathogenesis of the disease led to the discovery of a new type III hypersensitivity reaction mechanism originating in complement C5a that differed from the conventional one.
A real time imaging system for analysis
When we published our findings in the journal Science Immunology in 2017, the American Association for the Advancement of Science (AAAS) reported on it as a highlight the very same day, under the title “Tracking movement of immune cells identifies key first steps in inflammatory arthritis.” The reaction was such that it attracted the attention of around 30 media outlets overseas.
Whereas conventional in vivo imaging involves looking at cell dynamics, we succeeded in building a new kind of in vivo imaging system that analyzes the dynamics of chemotactic factors in tissue in real time. By that stage, we knew that there were two types of chemotactic factor receptors: classical receptors, which transmit signals, and atypical receptors, which do not. Unlike classical receptors, whose functions are comparatively well-understood, there was much we did not know about the functions of atypical receptors in the body. In particular, we had no idea how they functioned in inflammation.
The findings from our 2017 study had gone as far as revealing that C5a produced in joint tissue is transported to the vascular lumen and is deposited on the vascular endothelial cells, but we did not know what the transport mechanism was. Accordingly, we made full use of our in vivo joint imaging system and real-time analysis of chemotactic factor dynamics to investigate the mechanism via which C5a is transported from tissue to the vascular lumen.
As a result, we discovered that knocking out the atypical complement receptor C5aR2 inhibits the transport of complement C5a into blood vessels. In other words, by transporting C5a, the atypical complement receptor C5aR2 induces intravascular adhesion mediated by the classical complement receptor C5aR1.
In Science Immunology in 2019, we published the world’s first demonstrating a type III hypersensitivity reaction mechanism differing from the conventional one, an atypical complement receptor serving as a transport mechanism for chemokines and complement molecules, and inflammation caused by the interaction of an atypical and a classical complement receptor. Our 2017 and 2019 findings became the subject of a special feature on high-impact research in the same journal.
Diseases do not only affect joints, of course. If we could apply in vivo imaging to other organs to visualize them, we would be able to shed light on the movement of cells. At present, along with a team of staff at St. Marianna University, I am working on the visualization of various diseases, including those affecting such organs as the brain, heart, skin, kidneys, liver, and intestines. Hardly anywhere else in the world is a single laboratory able to create a multi-organ in vivo imaging system (Figure 3).
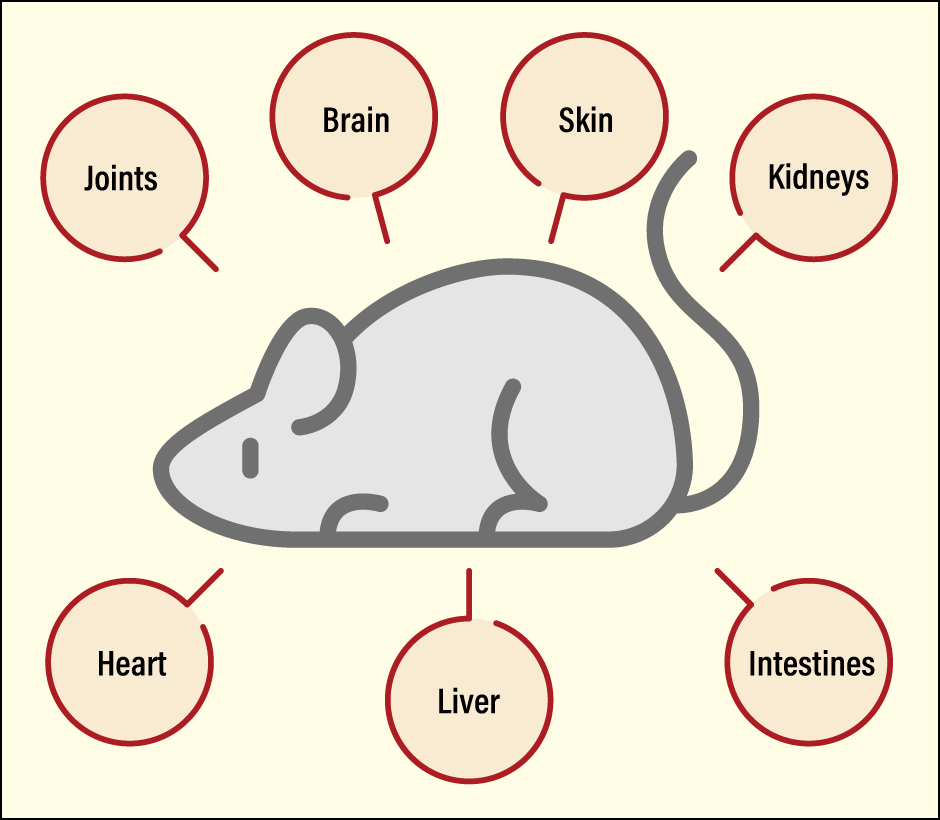
Figure 3. The imaging system used at Miyabe’s laboratoryMiyabe has built an in vivo imaging system that allows researchers to look at not only joints, but also many other organs, using mice. This system has rendered visible the movement of cells in multiple organs.
If the mechanism differs from one organ to another, there should be specific treatments for each one, and we are investigating what they might look like. However, these studies are still only being conducted in mice. Naturally, we want to look inside the human body, but using fluorescent labels in living people and exposing them to lasers for the purpose of observation is both technically and ethically impossible.
The question of how to overcome these issues is a challenge for us going forward. For my part, while I would like to somehow create a single drug targeting the cell migration mechanism, it is unlikely that we will be able to achieve this overnight. Accordingly, our goal for the time being is to ensure the findings we have obtained by visualizing diseases reach a wide audience, so that researchers across the globe can achieve further advances. I hope that drugs for these conditions will be developed a century or two from now. My own mother suffers from rheumatoid arthritis and it was seeing her struggling with the pain even when I was a child that made me want to become a physician specializing in rheumatology and connective tissue diseases. While my main focus today is basic research, I have not forgotten how I felt back then and hope to continue engaging in basic research that will be of use in clinical settings.
(Figures courtesy of Yoshishige Miyabe)