Platelets are a type of blood cell that stops bleeding. However, they are also involved in the pathologies of arterial thrombus formation. Although there are various causes of thrombocytopenia, inflammatory stress, such as viral or bacterial infection, rapidly decreases platelet production. Studies using mice have shown that inflammatory platelets are produced by a pathway different from normal under inflammatory stress. Furthermore, scientists have raised the possibility that a decline in platelet count and the production of inflammatory platelets may contribute to the sequelae of COVID-19 infection (long COVID).
Special Feature 1 – The Mechanisms and Functions of Blood Inflammatory platelet: unraveling the mystery of inflammation and sudden drops in platelet count
composition by Yumi Ohuchi
illustration by Rokuhisa Chino

Taisuke Kanaji
Institute Investigator, Department of Molecular Medicine, Scripps Research
After graduating from the Faculty of Medicine at Kyushu University in 1990, he worked at the First Department of Internal Medicine and the Department of Clinical Chemistry and Laboratory Medicine at Kyushu University Hospital, The Scripps Research Institute in the U.S., the Department of Biomolecular Sciences at Saga Medical School (now Saga University), the Division of Hematology (now the Division of Hematology and Oncology) at Kurume University School of Medicine, and the BloodCenter of Wisconsin (now Versiti) in the U.S. He returned to The Scripps Research Institute in 2013 and took up his current position in 2018. His main research topics are the analysis of inflammatory platelets using viral infection models and the identification of mechanisms of infection-induced autoantibody production.
All blood cells, including platelets, are created by differentiating hematopoietic stem cells in the bone marrow. Platelets are produced from megakaryocytes that have undergone differentiation from hematopoietic stem cells. Unlike in ordinary cells, the DNA in a megakaryocyte is replicated without cell division, so it matures into a polynuclear cell that is the largest cell found in bone marrow. A megakaryocyte extends platelet precursors (proplatelets) toward blood vessels; when the proplatelet breaks off, platelets are released into the blood vessel (Figure 1).
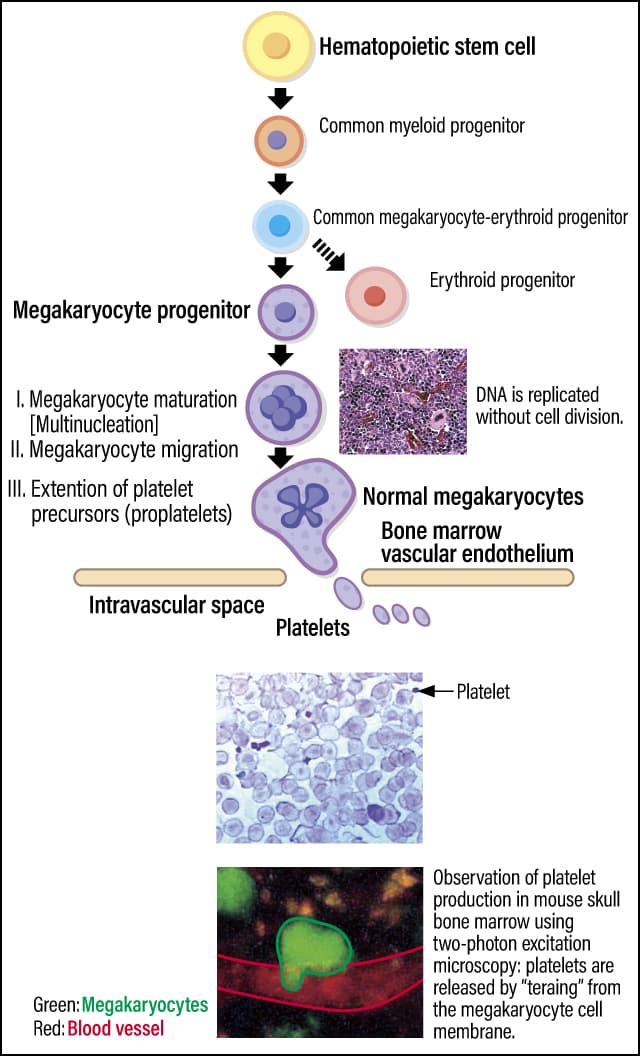
Figure 1. Platelets and their production mechanismPlatelets are produced from the cell membrane of polynuclear cells called megakaryocytes, which are differentiated from hematopoietic stem cells in bone marrow. Although megakaryocytes are the biggest cells in bone marrow, platelets are the smallest blood cells.
Therefore, the platelet becomes a small cell measuring 2-4 µm without nucleus. Scientists estimate that it takes 1-2 weeks for platelets to be produced from hematopoietic stem cells in humans and mice; once they have reached the end of their lifespan of around 10 days, they are disposed of in the spleen or, very occasionally, in the liver.
One of the most important physiological functions of platelets is adhering to the site of blood vessel injuries to stop bleeding. It is thanks to platelets that bleeding spontaneously stops. In addition, granules inside platelets store numerous growth factors that support tissue repair, and releasing these factors facilitates wound healing.
Inflammatory stress can also cause sudden decreases in platelet counts
While platelets have life-sustaining functions, they also contribute to pathologies involving the formation of arterial thrombi, such as heart attacks and strokes (Figure 2). When vascular endothelial cells in the arteries are damaged by factors such as high blood pressure, high cholesterol levels, smoking, and diabetes, the connective tissue (collagen fibers) beneath the vascular endothelium will be exposed. In such a situation, a protein called von Willebrand factor (VWF), secreted by vascular endothelial cells, binds to the exposed collagen fibers. As VWF normally flows through the bloodstream in a folded-up form, it does not bind to platelets. However, once VWF binds to collagen and unfolds, it captures platelets via a receptor called GPIbα, a platelet membrane protein. The activated platelets gather to form a thrombus, resulting in hemostasis and repair of the injured site. However, if the thrombus grows too large, it can block the blood vessels. In other words, hemostatic and arterial thrombus formation is a double-edged sword, which means appropriate activity levels and platelet count are crucial.
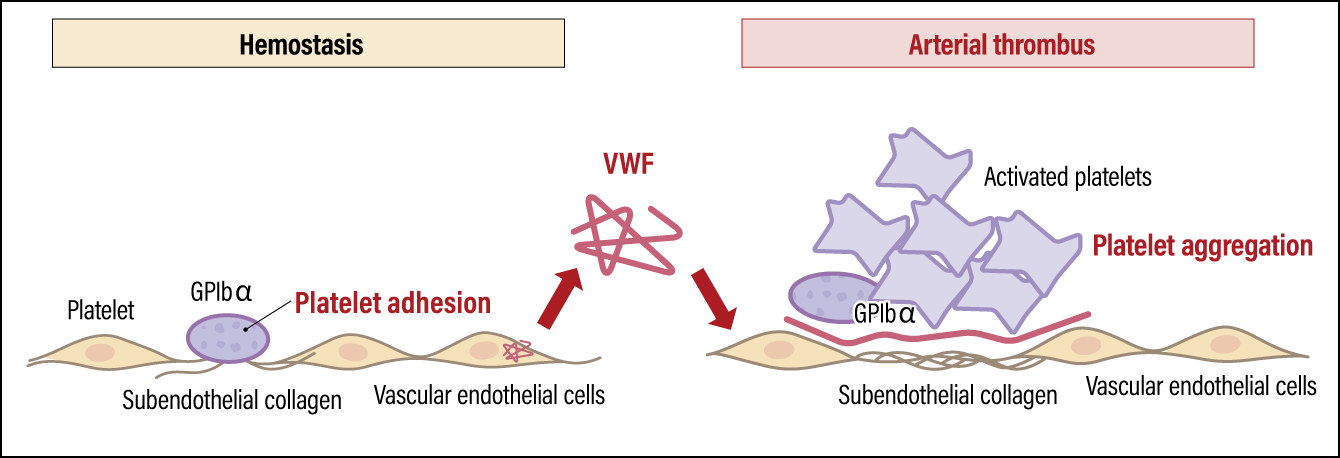
Figure 2. Platelet functions and pathologyPlatelets are essential to hemostasis, but they can also cause arterial thrombi. A platelet membrane protein called GPIbα plays a key role in both hemostasis and thrombus formation.
A decline in the number of platelets with hemostatic functions can be life-threatening. A variety of pathological conditions can cause a decrease in platelet count, including the production of autoantibodies (antibodies against components of one’s own body) against platelets and the side effects of anticancer drugs. Notably, inflammatory stress in the form of viral and bacterial infections can also cause sudden decreases. Our research has focused on this point.
One of our studies was conducted using lymphocytic choriomeningitis virus (LCMV) in mice: in one group, the infection was transient, and the virus was cleared within two weeks (acutely infected mice), while the other group of mice had a chronic infection that persisted from at least three months (chronically infected mice). While the house mouse (Mus musculus) is the natural host of LCMV, the virus is also transmissible to humans and is known to be a zoonosis that causes choriomeningitis.
When both groups of mice were infected with LCMV, they followed the same course, with a rapid decline in their platelet counts followed by recovery. However, after their infection, we found that a protein called Sca-1, which is not expressed in normal platelets, was expressed on platelets produced during inflammation. Sca-1 is a distinctive marker expressed on the surface of hematopoietic stem cells in mice.
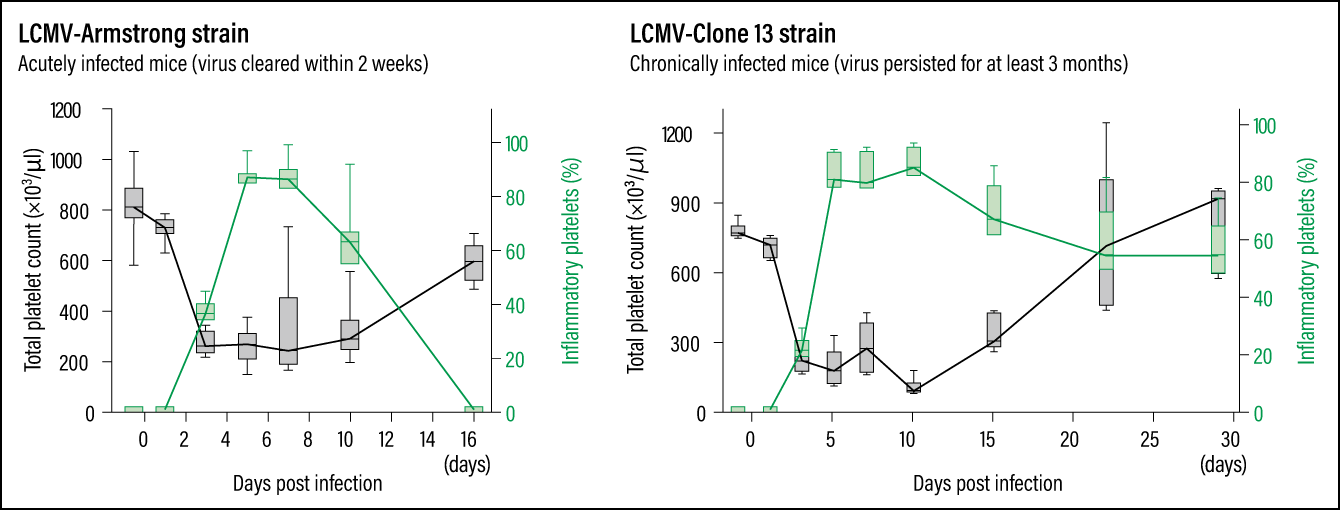
In other words, platelets generated during infection-induced inflammation (inflammatory platelets) appear to differ from normal platelets. Looking at changes in inflammatory platelets, we found that in the acutely infected mice, production of inflammatory platelets began to be induced on the third day after infection and accounted for most of the platelets in the bloodstream. Their numbers started falling on the eighth day and they disappeared about two weeks after infection. In chronically infected mice, inflammatory platelets were produced after infection, just like in acutely infected mice. Although a gradual decline in these cells was observed after 10 days of infection, we found that inflammatory platelets persisted and still accounted for about half of all platelets after a month (Figure 3). It is thought to be due to the persistent viral infection causing ongoing inflammation.
 Compiled based on Morodomi Y, et al: Proc Natl Acad Sci USA. 2022 Nov 29;119(48):e2212659119.
Compiled based on Morodomi Y, et al: Proc Natl Acad Sci USA. 2022 Nov 29;119(48):e2212659119.
Figure 3. Viral infection and stressAs indicated by the green lines, in the acutely infected mice, the production of inflammatory platelets began to be induced on the third day after infection; inflammatory platelets then began to decline on the eighth day and disappeared within about two weeks. In the chronically infected mice, on the other hand, although a gradual decline can start on the 10th day, inflammatory platelets still made up about half of the total platelet count a month after infection.
Now, the question is, why are inflammatory platelets produced? A rapid decline in platelet count increases the risk of hemorrhage. However, as stated earlier, it takes a week or two for normal platelets to be produced via the usual production pathway, so the body cannot respond quickly. Therefore, it is conceivable that a separate platelet production pathway exists, which increases the platelet number immediately after infection to avert a critical situation.
This pathway involves our immune system, especially the innate immunity that protects us from pathogens and other foreign substances. Innate immunity works as follows: when a pathogen (bacterium or virus) or other foreign substance invades the body, Toll-like receptors (TLRs) on immune cells rapidly detect it and produce interferons to attack the pathogen and swiftly defend the body.
Inflammatory platelets increase the risk of thrombosis.
Our research has brought to light the following platelet production pathway. When the body is stressed due to a viral infection, innate immunity is activated via TLR3/7/8. This results in the production of type I interferons, which play a core role in eliminating viruses; Type I interferons stimulate hematopoietic stem cells to activate hematopoiesis (blood formation), which is biased toward the production of megakaryocytes, the precursors to platelets. Thus, inflammatory platelets are rapidly produced (Figure 4). Interestingly, while type I interferons inhibit normal hematopoiesis, our study showed a stimulating action in megakaryocyte-biased hematopoiesis.
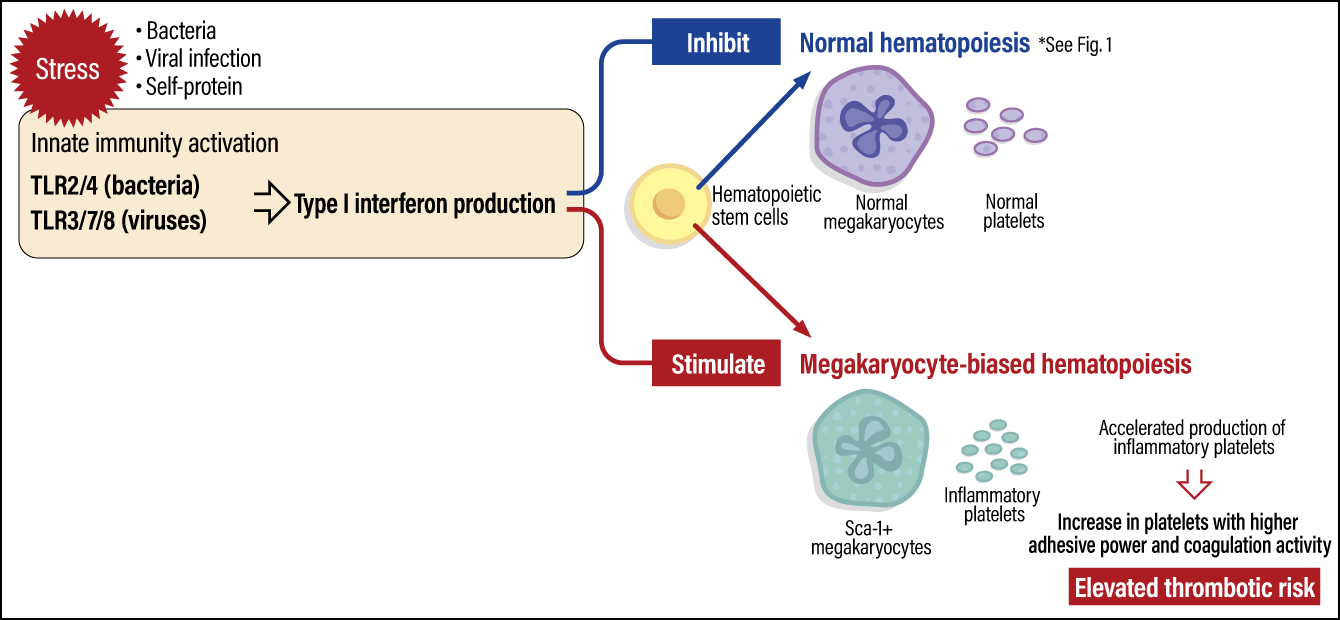 Compiled based on Morodomi Y, et al: Proc Natl Acad Sci USA. 2022 Nov 29;119(48):e2212659119.
Compiled based on Morodomi Y, et al: Proc Natl Acad Sci USA. 2022 Nov 29;119(48):e2212659119.
Figure 4. Inflammatory platelet production pathwaysWhen the body is subjected to stress due to a viral infection or the like, the activation of innate immunity activates blood formation biased toward megakaryocyte production, resulting in the production of inflammatory platelets, which swiftly defend the body. While inflammatory platelets have an increased hemostatic effect due to their high adhesive power and coagulation activity, they also elevate the risk of thrombus formation.
Our research has also shown that inflammatory platelets function differently from normal platelets. Inflammatory platelets exhibit higher adhesion and coagulation capacity than normal platelets. Accordingly, while they can efficiently control bleeding and prevent the contents of blood vessels from leaking out, they also increase the risk of thrombosis. With regard to the novel coronavirus COVID-19, the high incidence of thrombosis has attracted much attention, and we assume that inflammatory platelets are considered one of the causes of thrombosis associated with viral infection.
Another problem connected with COVID-19 is the sequelae (aftereffects) called long COVID, which is characterized by ongoing symptoms including exhaustion and fatigue, breathing difficulties, muscle weakness, cognitive impairment including poor concentration and impaired memory, and disturbance of the senses of taste and smell. Highly interesting research concerning the involvement of platelets has been reported in this area, as well. This study showed that type I interferons are secreted as a result of persistent infection of the gut by SARS-CoV-2, resulting in chronic inflammation and decreased absorption of the amino acid tryptophan from the gut. Serotonin is made from tryptophan, and impaired absorption of tryptophan leads to reduced serotonin. Serotonin is a neurotransmitter that regulates mood, emotions, sleep, and appetite, among others and is thought to have a link to depression. The authors concluded that this lack of serotonin could be a factor contributing to the neurological symptoms and cognitive dysfunction associated with long COVID.
Serotonin breaks down rapidly in the blood, but platelet granules retain it, maintaining its concentration. This study shows that reduced platelet count and the hypercoagulability of platelets produced in chronic inflammation are involved in serotonin deficiency. Our research also revealed that inflammatory platelets have a lower capacity for serotonin uptake than normal platelets, resulting in a reduced serotonin level stored in the granules. Therefore, we believe that the production of inflammatory platelets and their reduced capacity for serotonin storage may contribute to long COVID.
We experienced a valuable case related to thrombocytopenia (low platelet count) during the COVID-19 pandemic. The patient was seen at a university hospital in Japan with little red spots (petechiae) on their arms and bleeding from her oral mucosa following her second COVID-19 vaccination. Blood tests revealed that the patient’s platelet count had decreased to an unmeasurable level. It is known that even in undamaged blood vessels, platelets adhere to capillary endothelial cells and play an essential role in preventing blood from leaking out of the vessel. Accordingly, the sudden drop in the patient’s platelet count caused small hemorrhages due to the leakage of blood from the capillaries.
Hoping to assist in the diagnosis of long COVID
In collaboration with a university hospital, we found that the patient had produced autoantibodies against the GPIbα on platelets. These autoantibodies temporarily eliminated platelets from the patient’s blood, causing bleeding episodes. As described above, GPIbα is an essential protein for platelets to bind to VWF and carry out their hemostatic functions. We have previously shown that administration of the GPIbα antibody rapidly decreases platelet counts and produces inflammatory platelets in mice.
In this patient’s case, the COVID-19 vaccination triggered the production of autoantibodies against GPIbα and a sharp fall in her platelet count. However, the phenomenon was temporary, and the platelet count returned to normal after a few days. There have been reports of various autoantibodies being produced in patients infected with COVID-19, so one would presume that similar transient thrombocytopenia might have occurred in some of these cases after COVID-19 vaccination. In this case, platelets increased quickly just a few days after the platelet decrease, suggesting the involvement of inflammatory platelet production.
As described above, inflammatory stress by viral or other infections likely causes immune system dysregulation, inducing inflammatory platelets, with functions and production mechanisms distinct from normal platelets. These inflammatory platelets may contribute to various pathological conditions and post-infection sequelae. Sequelae following an infection are particularly difficult to diagnose, as with long COVID. Many patients suffer not only physical distress but also social burdens due to the lack of understanding from those around them. As such, if blood tests could detect the production of inflammatory platelets, it would be useful for assisting in rapid diagnosis. A specific marker for inflammatory platelets similar to Sca-1 has not yet been discovered in humans. Nevertheless, one would presume that a protein similar to Sca-1 exists in the human body, so we are working to identify it.
On a different note, GPIbα, which plays a vital role in thrombus formation, is an ideal target for developing drugs to inhibit thrombosis. However, as blocking the functions of GPIbα also impairs normal hemostatic functions and raises the risk of hemorrhage, so far, no antithrombotic drugs targeting GPIbα have been developed yet. In addition, the interaction of VWF and GPIbα is highly species-specific, so verification in animal research is challenging. We have already established genetically modified mice in which these molecules have been humanized, and we hope to contribute to the development of drugs that inhibit excessive thrombus formation, without impairing hemostatic functions.
(Figures courtesy of Taisuke Kanaji)