Organisms exert effects on each other as they evolve. Similarly, microorganisms have interacted with other organisms over the course of their survival. For example, the gut microbiome, which is composed of a diverse array of microbes, coexists with humans and virtually all other animals, and is involved in the biological activity of the host. Human microbiome is also greatly influenced by the host human. Accordingly, factors that are the root cause of dysbiosis —— changes in the gut microbiome —— have come under the spotlight. Shedding light on the mechanism would lead to more effective preventive measures and treatment.
Special Feature 1 – Forefront Research into Gut Bacteria Factors shaping the gut microbiome crucial for our health and disease
composition by Yumi Ohuchi

Masahira Hattori
Adjunct Researcher, Waseda University / Specially Appointed Professor, Faculty of Medicine, Juntendo University
After successfully completing a doctoral program at Osaka City University Graduate School of Engineering in 1979, he became Researcher Associate in the U.S. at the Scripps Research Institute and the University of California San Diego. His subsequent positions include Associate Professor at the Human Genome Center of the Institute of Medical Science, the University of Tokyo; Professor at Kitasato Institute for Life Sciences, Kitasato University; and Professor at the Graduate School of Frontier Sciences, the University of Tokyo (now Professor Emeritus). In 2015, he was Professor at the Faculty of Science and Engineering, Waseda University (now Adjunct Researcher). He became Team Leader at RIKEN Center for Integrative Medical Sciences in 2017 (now Senior Visiting Scientist). In 2023, he became Specially Appointed Professor at Juntendo University Faculty of Medicine.
Human microbiome is composed of around 40 trillion microbial cells, exceeding the 30 trillion cells of the human body. In other words, humans could be described as superorganisms united with the genetic information of humans and microbes. Accordingly, analysis of not only human genes, but also microbial genes in the human body is important to deeply understand us. Many studies of the microbiome have brought to light their close relationship to our physiological state and, consequently, health and disease.
Human gut microbiomes are largely diverse between countries
The microbiome is truly diverse: The number of the microbial species and their relative proportions vary from one part to another sites across the human body —— for example, between the skin, the oral cavity, the small intestine, the colon, and the stomach —— and there are also the differences between individuals. Moreover, the microbiome undergoes fluctuations throughout a person’s life. A recent article stated that the fetal environment in the uterus is sterile; it is only at the moment of birth that the baby is exposed to various microbes in the environment, such as the mother’s birth canal and the delivery room. However, it is thought that most of such early microbes are not major members establishing the microbiome in the oral cavity or gut at random, but rather that human-originated microbes that are selected over the course of a long evolutionary process take up permanent residence.
The gut microbiome is thought to be almost fully formed by about the age of 12, following changes in individual lifestyle and diet, including weaning off breast milk. While there are tolerant changes in the gut microbiome in infancy, it is maintained in a relatively stable state in adulthood, then later gradually changes as we age. Aside from the high degree of diversity between individuals, there are also differences between the sexes, with females host to a larger number of gut microbial species than males–specifically, female gut microbiome diversity is around 20% higher than the male level. While the reason for this is unclear, it is presumed to be related to longevity, given that women worldwide have higher life expectancies than men.
Our study showed that the species composition of the human gut microbiome also differs a great deal from one country to another as well as between individuals. In this study, we used metagenomic sequencing to compare the gut microbiomes of 861 healthy adults from 12 countries: Japan, China, the U.S., Peru, Venezuela, France, Austria, Denmark, Sweden, Spain, Russia, and Malawi.
- *1 Metagenomic sequencing: An analytical technique that involves directly and comprehensively sampling genomic information in microorganisms, without the need for culturing them.
The analysis of 106 Japanese individuals revealed a high inter-individual diversity in the composition, and found the high relative abundance of Bifidobacterium, Blautia, and Bacteroides in this dataset. Additionally, Bifidobacterium and Blautia were also highest in relative abundance among all 12 countries. One factor related to the high abundance of Bifidobacterium in the Japanese samples is milk consumption. This is because, compared with people from Western countries, most Japanese people secrete less of the lactase enzyme that breaks down the lactose in milk, so lactose reaches the gut without being broken down and serves as food for Bifidobacterium. It is thought likely that this abundant supply of food causes the Bifidobacterium population to grow.
The uniqueness of the Japanese gut microbiome
In the study of 861 healthy adults from 12 countries, we also compared the genes in the gut microbiomes between Japanese and individuals from 11 other countries for investigating functions that are significantly different between them. From the comparative analysis, we found that the distinctive functional features of the Japanese gut microbiome were a high carbohydrate metabolism capacity, a low abundance of flagellated microbes, and few genes associated with repair. Carbohydrates are metabolized to produce short-chain fatty acids, carbon dioxide, and hydrogen as end-products, of which short-chain fatty acids such as acetate and butyrate serve as nutrients for human cells, and hydrogen has an antioxidant action to suppress the oxidative stress. Accordingly, we can say that a high carbohydrate metabolism capacity is advantageous to the physiological state of humans. A low abundance of flagellated microbes means that an inflammatory immune response is less likely to occur, as humans experience immune responses to flagellar antigens in bacteria, particularly in pathogens. A low number of genes associated with repair is thought to signify that there is little genetic and cell damage in the gut. These characteristics might be related to the low BMI and high longevity of Japanese people compared with that of people in Western developed countries.
Also distinctive among the Japanese group was the fact that, of the hydrogen-metabolizing microbes, there was a high abundance of Blautia, but a low abundance of the archaeon Methanobrevibacter smithii. In hydrogen metabolism, M. smithii generates methane, while Blautia produces acetate, which suggests that the metabolic pathway of hydrogen in the gut differs in Japanese people from that in people from other countries. We know from existing studies that the level of methane in the exhaled breath of Japanese people is lower than that among people from other countries, so it is likely that the reason relates to this difference in the metabolic pathway of hydrogen by gut microbes.
Aside from this, we found that the gut microbiomes of around 90% of the Japanese individuals contained the gene for the enzyme porphyranase, which breaks down polysaccharides in green laver and wakame kelp, whereas this gene was found in fewer than 15% of the individuals from the other 11 countries. , The porphyranase gene is ubiquitous in marine bacteria. It is thought that, because Japanese food culture involves the consumption of large amounts of raw fish and marine products, the porphyranase gene from marine bacteria has undergone horizontal gene transfer to human gut bacteria, which are persist in many of Japanese people with food culture of the marine product.
- *2 Horizontal gene transfer: A phenomenon in which genes are transferred between microorganisms, either within the same species or between different species, without being transferred between generations.
When we looked at the relative abundance of microbial genera detected in each country, we found they varied from one country to another, as stated earlier, and interestingly, it was found that the countries could be divided into three groups: a group with a high abundance of Bifidobacterium and Blautia, which includes Japan; a group in which Bacteroides was prevalent; and a group in which Prevotella was predominant (Figure 1). The other countries in the group including Japan are Sweden, France, and Austria, which at first glance would seem to have no points in common with Japan. The same applies to the group with a high abundance of Bacteroides, e.g., a high similarity between the US and China, while members of the group with a high abundance of Prevotella have in common the fact that they are developing countries.
 Compiled from Nishijima S, et al. DNA Res, 23: 125-133, 2016.
Compiled from Nishijima S, et al. DNA Res, 23: 125-133, 2016.
Figure 1. Microbial compositions in 12 countriesCountries were sorted into three groups, based on the results of hierarchical cluster analysis (clustering together data groups that are mathematically similar) of similarities in each country’s microbial composition.
Dietary habits are the primary thing that springs to mind as a factor affecting the gut microbiome. However, even thinking in simplistic terms, one could not say that, for example, Japan and France had the same dietary habits, nor China and the U.S. Even when we used a database of global dietary information to analyze the data, we were still unable to explain the similarities or dissimilarities of each group based on diet and the gut microbiome. This suggested the existence of a factor other than diet that substantially affects the gut microbiome structure. When we conducted further investigations that also incorporated apparently unrelated factors, such as each country’s land area and latitude, the amount of antibiotic usage stood out. We conducted an association analysis of the gut microbiome data in our study and a database of antibiotic usage, whereupon we found that the countries where antibiotic usage was relatively high were aggregated in the group in which Bacteroides was most abundant; countries where usage was relatively moderate were in the group where Bifidobacterium and Blautia were prevalent; and countries with little antibiotic use were in the group where Prevotella was predominant (Figure 2).
 Compiled from Nishijima S, et al. DNA Res, 23: 125-133, 2016.
Compiled from Nishijima S, et al. DNA Res, 23: 125-133, 2016.
Figure 2. Antibiotic usage and microbial compositionThis indicated the possibility that antibiotic usage has a bigger impact on gut microbiome composition than diet. The range accounted for by Bacteroides in Russia was smaller than in other countries in the same group, while antibiotic usage there was moderate.
In addition, we discovered that Bacteroides levels were higher in countries with higher antibiotic usage —— in other words, the figures showed a positive correlation between them, which was particularly high in the case of penicillin. The results of this analysis led us to think that antibiotic usage has a more substantial impact on the gut microbiome than diet. We can easily imagine the magnitude of this impact when we consider that, although antibiotics are specifically intended for treating infectious diseases, they kill not only pathogens, but also gut microbes and others. In the U.S., where antibiotic usage is said to be high, some studies have reported results indicating a positive correlation between obesity rates and antibiotic use.
Medications have a larger impact than diet
What I want to clarify is which factors affect the human microbiome and which factors have a substantial impact on it. Possible internal factors include human physiological states in the form of age, sex, circadian rhythm, and pregnancy, as well as host’s genetic background and diseases, while living environment, lifestyle, dietary habits, and medications being taken are among the possible external factors. In particular, various common diseases are known to be linked to the altered gut microbiome, including metabolic diseases such as obesity, diabetes, metabolic syndrome, renal failure, and heart failure; immune and inflammatory system diseases such as inflammatory bowel disease, irritable bowel syndrome, rheumatoid arthritis, arteriosclerosis, allergies, and cirrhosis; neurological and psychiatric disorders such as autism spectrum disorder, multiple sclerosis, dementia, and Parkinson’s disease; and cancers such as colon cancer, liver cancer, and pancreatic cancer. We know that, compared with those of healthy individuals, the gut microbiomes of patients with many of these diseases characteristically differ in microbial species and compositions. The word “dysbiosis” is used to describe such differences in the gut microbiome from that of healthy individuals.
This, then, was the backdrop to a project in which we worked with a team led by Dr. Naoyoshi Nagata of Tokyo Medical University to build a database that integrated information about the gut microbiomes with individual metadata such as diet and exercise habits, diseases, and medications of around 4,200 Japanese subjects. This database may be the first and largest gut microbiome database in Japan at this time, also one of the world’s largest. In this project, we identified 1,773 gut bacteria at the species level, 10,689 genetic functions of gut bacteria, and 403 antibiotic resistance genes by metagenomic sequencing and analysis of fecal DNA.
In the association analysis, we found that medication has the biggest impact, followed by diseases; physical information, such as age, sex, and BMI; dietary habits; and lifestyle (smoking and alcohol), and exercise has the lowest impact among various metadata (Figure 3). Also worthy of note is that the effect of medication is more than three times greater than dietary habits, lifestyle or exercise.
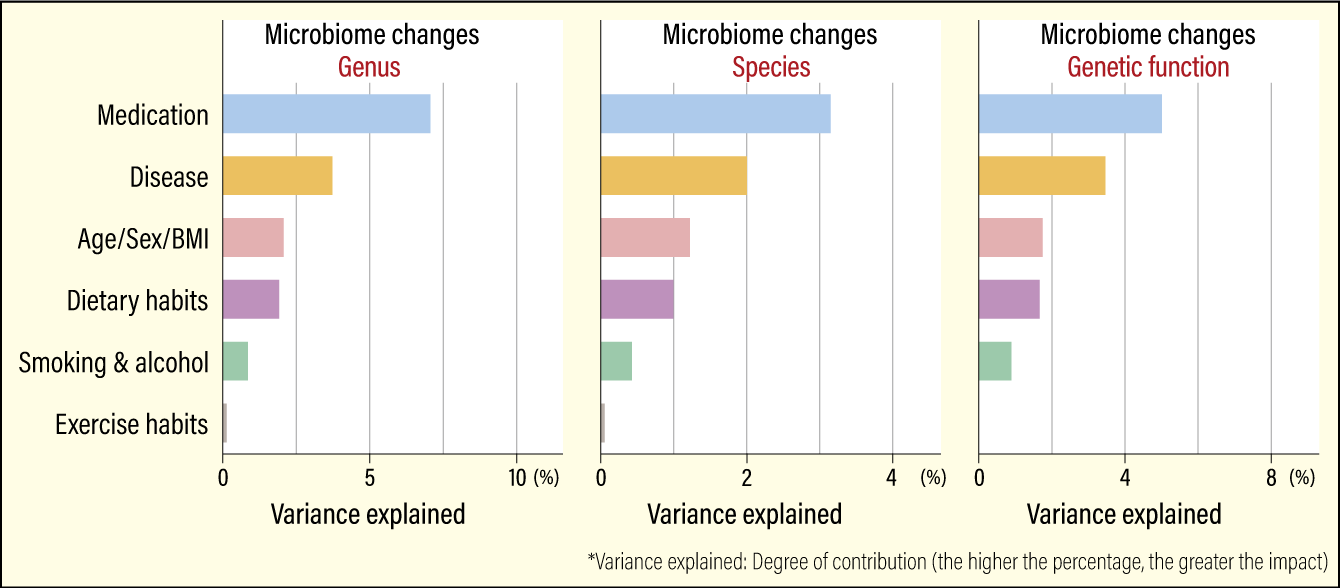 Modified from Nagata N, et al. Gastroenterology, 163:1038-1052, 2022 Oct.
Modified from Nagata N, et al. Gastroenterology, 163:1038-1052, 2022 Oct.
Figure 3. Factors affecting the gut microbiomeThe effect of medication is more than three times greater than that of dietary habits, lifestyle, or exercise. We also obtained the similar results concerning the impact of these affecting factors on the gut microbiome at the genus, species, and genetic function levels.
In addition, people who used a higher number of medications concurrently had lower gut microbiome diversity, with an increase in the number of pathogens that cause opportunistic infections. Opportunistic infections are infections caused by pathogens that are not pathogenic under normal circumstances, but which exhibit pathogenic properties when the host’s immune function is imbalanced.
The need to consider factors associated with the gut microbiome
When we examined which medications against disease had a substantial impact on the gut microbiome, we found that the biggest impact came from alimentary tract drugs, followed in descending order by diabetes drugs, antibiotics, antithrombotics, cardiovascular system drugs, nervous system drugs, anti-cancer drugs, musculoskeletal drugs, genitourinary drugs, and other drugs (respiratory system drugs and traditional Chinese medicine). The most distinctive characteristic to emerge was that using a proton-pump inhibitor (PPI), which is a drug that inhibits the secretion of stomach acid, and an alpha-glucosidase inhibitor (α-GI), which is a diabetes drug. The effect of both α-GIs and PPIs is to reduce blood glucose level by slowing down the decomposition of dietary polysaccharides producing sugar in the gastrointestinal tract. We found that both drugs significantly reduced gut microbiome diversity and changed the microbial composition in cases where those were administrated. The combined use of both drugs more strikingly altered the gut microbiome than either single use.
Furthermore, we compared fecal samples from patients before and after the administration of PPIs to examine whether the drug was responsible for changes in their gut microbiome or whether those who took the drug had already experienced a change in their gut microbiome. We found that the abundance of Lactobacillus and Streptococcus increased in patients who started to use PPIs, in concurrent with increase in that of opportunistic pathogens, such as Enterococcus faecalis and Streptococcus pneumoniae. After the patients halted the use of PPIs, the abundance of these microbial species was mostly restored, which suggests that the effect of PPIs on the gut microbiome is prominent but temporary on taking them.
Thus, unlike the food we eat every day, medications have powerful effects on the gut microbiome more than we thought. There is, therefore, a need to consider effects of drugs on the gut microbiome when prescribing and developing them.
As I have already stated, it is plain to see that the gut microbiome and particularly its dysbiosis is connected to human disease and health. Dysbiosis of the gut microbiome is profoundly associated with disease onset and sometimes its prognosis. Consequently, once we shed light on the detailed mechanism behind it, we will be able to develop more effective treatment and prevention of diseases by controlling the gut microbiome.
However, many things remain unknown about the gut microbiome, which consists of not only bacteria, but also viruses (phages) that infect bacteria, and small DNA molecules called plasmids present inside bacteria, all of which also possess many genes for distinct functions from the bacterial chromosomes. Methods for analyzing metagenomes that allows us to examine dark matters in the gut microbiome have recently been developed. The trigger factors of disease onset might actually lie in these places that were hitherto hidden from us. I will continue working on microbiome research, in an effort to explore them.
(Figures courtesy of Masahira Hattori)